Protein-Interaktomik
Wer macht‘s mit wem?
von Mario Rembold, Laborjournal 11/2016
Um zu verstehen, wie ein Organismus funktioniert, reicht es nicht, seine Gene und Proteine nur zu kennen. Man muss wissen, wie genau die Proteine zusammenarbeiten. Insbesondere die „Interaktomik“ widmet sich der Suche nach solchen Protein-Protein-Interaktionen. Zum Beispiel mit Massenspektrometrie oder mit Yeast Two Hybrid-Methoden.
Jörg und Mia gehören zusammen, denkt der Kioskbesitzer gegenüber. Er sieht die beiden mehrmals täglich gemeinsam vor dem Haupteingang stehen. Dabei arbeiten sie in vollkommen verschiedenen Abteilungen der Firma und haben dienstlich nie direkten Kontakt. Was sie verbindet, sind allein die Zigarettenpausen. Die meiste Zeit des Arbeitstages verbringt Jörg im Büro mit Bernd. Die beiden sind ein eingespieltes Team, und sie treffen sich einmal pro Woche mit Hanna und Beate aus der Niederlassung in der Nachbarstadt. Bei diesen kurzen aber effektiven Meetings sind in den letzten Jahren Ideen entstanden, denen die Firma einen Großteil ihrer Umsätze verdankt. Norbert im Büro gegenüber kennt Hanna und Beate gar nicht persönlich und sieht immer bloß, wie Jörg und Bernd zusammenarbeiten.
Bioforscher vergleichen das Zusammenspiel der Proteine innerhalb einer Zelle gerne mit solchen sozialen Interaktionen. Unter anderem auch, weil Analogien wie diejenige zu Jörg und seiner Rolle in der Firma bereits sehr schön ein ganz allgemeines Problem aufzeigen: Es ist keineswegs trivial zu erkennen, welche Interaktionen tatsächlich wichtig sind und welche nicht. Je nach „Messmethode“ findet man vielleicht nur das „Raucher-Duo“. Um der Bedeutung des „Kreativ-Quartetts“ aber auf die Schliche zu kommen, muss man schon sehr genau hinschauen. Schließlich sind die Vier nur selten zusammen, obwohl dieser Kontakt äußerst wichtig ist für die Verkaufszahlen unseres fiktiven Unternehmens.
Umso mehr steht ein Forscher vor großen Herausforderungen, wenn er die Protein-Protein-Interaktionen einer Zelle und deren Bedeutung erfassen möchte. Weil das Ganze mehr ist als bloß ein Nebenschauplatz der Proteomik, hat sich für diese Disziplin ein eigener Name etabliert: die Interaktomik. Und das Gesamtnetzwerk aller Protein-Protein-Interaktionen eines Organismus’ nennt man folglich das „Interaktom“.
Erstmal mit der Schrotflinte
Zur Entschlüsselung solcher Interaktome setzen viele Proteom-Forscher auf die Massenspektrometrie – so auch Matthias Selbach, Leiter der Arbeitsgruppe „Proteome dynamics“ am Max-Delbrück-Centrum für Molekulare Medizin in Berlin. „Wenn ich weiß, mit wem ein Protein interagiert, dann weiß ich oft auch, was es macht“, unterstreicht Selbach die Bedeutung der Interaktomik. „Darin liegt häufig der Schlüssel zum funktionellen Verständnis eines Proteins.“
In vielen Fällen beginnt die Identifizierung mit einem Protein-Verdau, um Peptide zu erhalten, die zwischen sieben und dreißig Aminosäuren lang sind. „Shotgun Proteomics“ nennen die Proteinjäger ihre etwas rabiate Probenanalyse. Im Massenspektrometer lassen sich die Peptide dann anhand ihrer Molekülmassen grob unterscheiden. Diese wiederum verrät, aus welchen Aminosäuren das Peptid bestehen könnte. Natürlich sind dann immer noch verschiedene Reihenfolgen der Aminosäuren möglich – weshalb das Massenspektrometer die Peptide anschließend fragmentiert und eine zweite Messung durchführt. „Unterschiedliche Peptide haben ganz charakteristische Fragmentierungsmuster“, weiß Selbach. „Zusammen mit der Masse des Peptids kann man dann recht zuverlässig identifizieren, um welches Peptid es sich handelt“. Im letzten Schritt verrät der Abgleich mit Proteindatenbanken schließlich, zu welchem Protein ein Peptid gehört.
Um nicht verschiedene Peptide derselben Masse zu untersuchen und ein bisschen Ordnung in den Molekül-Klumpatsch zu bringen, trennt man die Proben vor der Massenspektrometrie chromatografisch auf – heute Standard in den Laboren der Protein-Biochemiker. Dort ist die HPLC dann direkt an ein Massenspektrometer gekoppelt und übergibt die Proben automatisiert im Hochdurchsatz.
Köder auswerfen
Nicht selten ist der Wissenschaftler an einem ganz bestimmten Protein interessiert, nennen wir es Protein A. Er möchte wissen, welche anderen Proteine an A binden können. Mittels Immunpräzipitation kann man nun Protein A herausfischen und bekommt dann mit etwas Glück auch Proteine zu fassen, die an A haften. Protein A dient also als Köder oder „bait“, um an die noch unbekannten Interaktionspartner heranzukommen. Alles, was man braucht, ist einen Antikörper gegen Protein A, um dieses zusammen mit seiner „Beute“ in einer Probe anzureichern. „Der muss aber wirklich spezifisch und mit einer guten Affinität an mein Protein binden“, betont Selbach. Fehlt ein solcher Antikörper, kann man sich mit einem Tag behelfen: Man ergänzt Protein A um eine Domäne, gegen die man einen Antikörper zur Hand hat. Das Gen dieses Fusionsproteins bringt man ins Genom seiner Zelllinie oder eines Modellorganismus ein und kann damit nach Interaktionspartnern fischen und diese im Massenspektrometer identifizieren.
„Damit erhalte ich aber erstmal nur Massen und Intensitäten“, erklärt Selbach und kommt auf ein Problem zu sprechen: „Leider kann ich aus der Intensität eines Peptids nicht ohne weiteres auf seine Menge oder Konzentration schließen“. Quantifizierung im Massenspektrometer ist immer nur möglich, indem man Peaks desselben Moleküls miteinander vergleicht. Vielleicht findet man ein Protein B, das während der Mitose doppelt so häufig an Protein A klebt. Für eine Stöchiometrie – auf jedes Protein A kommen n Proteine B – braucht es außerdem noch absolute Referenzwerte. Die bekommt man, indem man zuvor Proben mit bekannten Konzentrationen der Peptide durchmisst.
Mal mehr, mal weniger Interaktion
Tatsächlich wird man für die allermeisten „interaktomischen“ Fragestellungen zumindest relative Quantifizierungen vornehmen. Denken wir an das Eingangsbeispiel mit Jörg und seinen Kollegen. Schaut ein Beobachter zufällig immer wieder mal hin, wird er dessen Bürokollegen Bernd als wichtigsten Partner finden. Erst über einen gezielten Vergleich mit „Messungen“ nur während der Pausenzeiten kann der Beobachter feststellen, dass auch Rauchpartnerin Mia eine Bedeutung in Jörgs Arbeitsalltag haben könnte.
Dasselbe gilt für Protein-Protein-Interaktionen in der Zelle. Gut möglich, dass ein Rezeptor nur selten mit seinem Liganden interagiert, dann aber ein wichtiges Signal weiterleitet. Umgekehrt kann es beim Lysieren der Zellen zu Interaktionen kommen, die in der lebenden Zelle keine Rolle spielen. „Man kann sicherlich sagen, dass alle Proteine irgendwie mit allen anderen Proteinen interagieren können“, meint Selbach. „Nur dürften die allermeisten Interaktionen physiologisch irrelevant sein“.
Somit sind es oft erst die Vergleiche zwischen zwei Zuständen, über die wichtige Partner entdeckt und mit einer biologischen Funktion in Zusammenhang gebracht werden können. „An Quantifizierung führt letztlich kein Weg vorbei“, bring es Selbach auf den Punkt. Solche Unterschiede auf elegante Weise und quasi „in einem Rutsch“ erkennen kann man mittels Stable Isotope Labeling with Amino Acids in Cell Culture – kurz: SILAC; eine Technik, die ursprünglich das Team um Matthias Mann entwickelt hatte, der heute am Max-Planck-Institut für Biochemie in Martinsried forscht (Mol Cell Proteomics 1(5): 376-86).
Auch Selbach und seine Kollegen nutzen SILAC für ihre Interaktom-Experimente. „Es ist ein Zellkultur-basiertes Verfahren, bei dem man essentielle Aminosäuren wie Arginin oder Lysin mit schweren Isotopen markiert“, erklärt Selbach. Diese Isotope sind stabil und nicht radioaktiv. Peptide mit diesen Aminosäuren haben dieselben chemischen Eigenschaften und auch die gleiche „Flyability“ im Massenspektrometer. Anhand ihrer Masse kann man sie aber unterscheiden.
Verräterische Masse
Nun kann man eine Säugerzelllinie auf einem Medium mit den markierten Aminosäuren wachsen lassen. In parallelen Ansätzen hält man die gleichen Zellen auf normalem Nährmedium ohne Isotopen-Label. Jetzt ändert man in der einen Versuchsreihe gezielt Bedingungen, die für die Protein-Protein-Interaktion interessant sein könnten. Zum Beispiel, indem man einen Wachstumsfaktor zugibt. Zum Lysieren der Zellen und für die folgende Immunpräzipitation gibt man die Proben dann zusammen. So werden beide Ansätze ab diesem Schritt exakt gleich behandelt. Im Massenspektrometer verraten sich die gelabelten Proteine des einen Ansatzes dann anhand ihrer höheren Masse; ist die Intensität einzelner Peptide der Interaktionspartner höher oder geringer als in der Kontrollprobe, dann hat man offenbar einen Parameter entdeckt, der die Protein-Protein-Interaktion beeinflusst.
Mit Hilfe von SILAC hat Selbachs Team beispielsweise Interaktionspartner der c-Jun N-terminalen Kinase (JNK) identifiziert (Mol Cell Proteomics 14(1): 50-65). Diese Kinase kommt in unterschiedlichen Splice-Varianten vor. JNK kann den Zelltod von Neuronen induzieren und spielt bei Differenzierungsprozessen und neuronaler Regeneration eine Rolle. Als Modell für neuronale Zellen hatten die Forscher eine PC12-Zelllinie der Ratte genommen. Sie wussten, dass der Nervenwachstumsfaktor NGF Differenzierungsprozesse initiiert, die über JNK vermittelt werden. Also verglichen sie die Interaktion zwischen JNK und anderen Proteinen mit und ohne NGF-Signal und fanden Sfpq und Nono als signalspezifische Interaktionspartner.
Die gute, alte Hefe
Neben der Massenspektrometrie gibt es aber auch molekularbiologische Methoden, um Protein-Protein-Interaktionen auf die Schliche zu kommen. Ein Klassiker der Proteomik ist hier das Yeast Two Hybrid-System (Y2H). Dabei kommt der Transkriptionsfaktor GAL4 zum Einsatz. Dieser hat in seiner Originalversion eine DNA-Bindedomäne sowie eine Domäne, die die Transkription der nachgeschalteten Sequenz anwirft. Nun kann man die GAL4-Domänen auf getrennten Genen in Saccharomyces unterbringen. Natürlich funktionieren die beiden halben GAL4-Proteine dann nicht mehr – der einen Version fehlt die Transkriptionsdomäne, die andere Version findet ihr Ziel auf der DNA nicht. Kloniert man nun jeweils eine Sequenz eines Kandidatengens hinter die beiden GAL4-Gene, erhält man zwei Fusionsproteine. Interagieren Domänen der beiden Kandidatenproteine miteinander, dann bringen sie dadurch auch beide GAL4-Domänen wieder zusammen. Es kommt zur Genexpression – und natürlich kann man dafür ein Reportergen einsetzen, das ein leicht messbares Signal generiert oder einen modifizierten Hefestamm überhaupt erst lebensfähig macht. (In letzterem Fall wachsen nur Kulturen mit interagierenden Proteinpaaren).
Y2H kommt in unzähligen Modifikationen in Saccharomyces zum Einsatz. Fusionsproteine mit einer der beiden GAL4-Domänen kann man auf einem Plasmid hinterlegen und bei Bedarf in Hefezellen einbringen. Und weil auch die Hefe-Genetik heute keine Hexerei ist, lassen sich so in überschaubarer Zeit massenhaft Plasmidbanken mit Kandidatengenen screenen; Zellen, die ein Reportersignal generieren, enthalten einen möglichen Interaktionspartner. „Der große Nachteil dieser Methode ist, dass sie nicht quantitativ ist“, merkt Selbach an. „Man bekommt eine binäre Antwort: Entweder ist das Reportergen ein- oder ausgeschaltet.“ In jeder Hefezelle treffen immer genau zwei Kandidaten-Proteine aufeinander. „Das kann man jetzt als Vor- oder Nachteil der Methode ansehen“, resümiert Selbach.
Erich Wanker sieht genau darin den besonderen Reiz des Y2H-Systems. „Wir messen immer nur die direkten Interaktionen“, schwärmt er. „In der Massenspektrometrie habe ich vielleicht einen größeren Komplex, sehe aber nicht, wer jetzt mit wem direkt wechselwirkt.“ Somit ist klar, dass es auf die Fragestellung ankommt: Wer ganze Proteinkomplexe finden will, wird auf Immunpräzipitation und Massenspektrometrie setzen. Wer hingegen nur an direkten Interaktionen interessiert ist, dürfte eher dem Hefemodell zugeneigt sein.
Bindet‘s auch im wirklichen Leben?
Wanker leitet am Berliner Max-Delbrück-Centrum für Molekulare Medizin die Arbeitsgruppe „Neuroproteomics“ und möchte ebenfalls wissen, wie Proteine zusammenarbeiten. Nun kann es passieren, dass ein Hefe-Screen sehr deutliche Protein-Protein-Interaktionen zutage fördert, die in der lebenden Säugerzelle niemals zu sehen sind. Vor allem dann, wenn man einfach blind Plasmidbanken durchscreent. Vielleicht sieht man, wie ein neuronenspezifisches Protein an einen Partner bindet, der nur im Zytosol von Leberzellen vorkommt. In einer Zelllinie eines ausgewählten Gewebetyps wäre solch ein irreführender Befund weniger wahrscheinlich. „Wir sehen Yeast Two Hybrid als ein lebendes Test-Tube“, betont Wanker an dieser Stelle. „Wenn wir Y2H einsetzen, interessiert uns erstmal, ob zwei Proteine überhaupt wechselwirken können“, ergänzt der Forscher. Dabei messe man zunächst bloß eine biophysikalische Interaktion. „Ob die biologisch relevant ist oder nicht, kann erst in weiteren Untersuchungen geklärt werden.“
Dank umfangreicher Transkriptom-Datenbanken tappt man bei der Interpretation von Y2H-Ergebnissen allerdings auch nicht völlig im Dunkeln. „Da schaut man natürlich nach, ob solch ein Protein überhaupt im Gehirn exprimiert sein kann“, erläutert Wanker im Hinblick auf sein Forschungsgebiet. Dabei kann Y2H auch helfen, krankhafte Mechanismen aufzuklären, die durch veränderte Aminosäuresequenzen eines Proteins begünstigt werden. Nämlich indem man nach Unterschieden zwischen mutierten und gesunden Varianten desselben Proteins sucht und dabei womöglich unterschiedliche Interaktionspartner findet.
„Die Interaktion mit CRMP haben wir unter anderem mithilfe von Y2H gefunden“, nennt Wanker ein Beispiel. Die Auswertung umfangreicher Neuroproteom-Daten hatte nämlich ergeben, dass das neuronenspezifische Protein CRMP1 der Bildung von Proteinaggregaten entgegenwirken kann, wie sie bei Chorea Huntington typisch sind (Genome Res 25(5): 701–13).
In Wankers Gruppe arbeitete Philipp Trepte während seiner Doktorarbeit auch an einem binären Nachweissystem, das in Säugerzellen eingebracht wird und für das ebenfalls zwei Fusionsproteine zum Einsatz kommen. Jedes trägt eine andere Luciferase, die jeweils durch Biolumineszenz einer charakteristischen Wellenlänge identifizierbar ist. Trepte konnte die Zellen dann lysieren und die interagierenden Proteine mit einem Antikörper gegen eine der beiden Luciferasen immunpräzipitieren. Dann verglich Trepte die Intensität beider Luciferase-Signale und konnte so auf das Verhältnis beider Proteine im interagierenden Komplex schließen. „Somit wird ein quantitatives Signal erzeugt“, erklärt Trepte und nennt einen weiteren Vorteil: „Man kann zum Beispiel die Effekte krankheitsrelevanter Mutationen auf eine Interaktion untersuchen.“ DULIP nennt sich diese Methode, für Dual Luminescence-Based Co-Immunoprecipitation Assay (J Mol Biol 427(21): 3375-88).
Auch wenn Interaktomik stark von der Arbeit an Zellkulturen geprägt ist, lassen sich natürlich auch ganze Gewebe massenspektrometrisch analysieren. Und Two-Hybrid-ähnliche Systeme sind mittlerweile nicht nur auf Hefe beschränkt, sondern man kann sie auch in Würmern, Fliegen oder Mäusen einsetzen. „Die perfekte universelle Methode, die alles abgreift, sehe ich aber noch nicht“, meint Trepte zum aktuellen Stand der Interaktomik. Somit darf man noch viele Weiterentwicklungen erwarten. Natürlich auch, was die Auswertung und Interpretation solcher Daten betrifft.
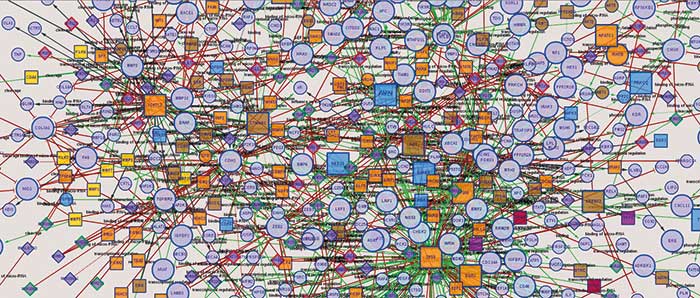
Kommen wir an dieser Stelle zurück zum Netzwerk der Protein-Interaktionen: Wer tritt mit wem in Kontakt? Marco Hein, inzwischen seit zwei Jahren an der University of California in San Francisco, hatte sich zuvor als Doktorand in der Gruppe von Matthias Mann am Martinsrieder MPI für Biochemie nicht bloß ein einzelnes Protein ausgeguckt und dessen Interaktionspartner gesucht, sondern wollte gleich ein gesamtes Netzwerk – das humane Interaktom – im Ergebnisteil seiner Doktorarbeit sehen. Daher führte er Screenings für mehr als eintausend Proteine in HeLa-Zellen durch, um jeweils deren Interaktionspartner zu finden. Aus seiner Doktorarbeit wurde schließlich ein Cell-Paper, an dem auch Zellbiologen aus Dresden beteiligt waren (Cell 163(3): 712-23).
Warum nicht alle auf einmal?
Am Anfang stand zunächst ein möglichst komplettes HeLa-Proteom. „Das haben wir absolut quantifiziert, um die Konzentrationen jedes einzelnen Proteins zu kennen“, blickt Hein zurück. Dann ging es weiter mit seinen rund tausend Beispiel-Proteinen, jedes davon mit einem Tag für die Immunpräzipitation, um Interaktionspartner zu finden. Das Referenz-Proteom ermöglichte Hein schließlich auch quantitative Aussagen zur Häufigkeit der Bindungspartner und zur Stöchiometrie der Proteinkomplexe.
Einen Teil des Netzwerks haben Hein und seine Kollegen im Paper grafisch dargestellt. Man sieht einige Cluster, in denen Proteine eng beieinander liegen, die auch funktionell zusammenarbeiten müssen. So gibt es eine Gruppe, in denen sich Proteine sammeln, die für das Cytoskelett zuständig sind – und in einem anderen Büschel vereinigen sich Proteine, die Prozesse rund um die Ribosomen steuern. Verbindungen innerhalb eines Clusters sind vergleichsweise stark. „In vielen Interaktomik-Experimenten findet man nur diese starken Interaktionen“, erklärt Hein und betont, dass man dabei viele isolierte Inseln bekomme, und eben kein zusammenhängendes Netzwerk. Es seien erst die schwachen Interaktionen, die diese Inseln miteinander verbinden. „Über eine Kette vieler schwacher Interaktionen kann in der Zelle auch ein funktioneller Zusammenhang entstehen“, schlussfolgert Hein aus den Daten.
Schwach aber wichtig
Unter „Stärke“ sei hier nicht unbedingt die Stabilität des jeweiligen Proteinkomplexes zu verstehen. „Dazu gab es viele Rückfragen der Gutachter zu den ersten Versionen unseres Papers“, erinnert sich Hein. „Denn dieser Begriff ist ja schon biophysikalisch besetzt – da sind wir dann mit unserer Wortwahl vorsichtiger geworden“. Denn eine „schwache“ Verbindung im Netzwerk muss nicht heißen, dass die Interaktion auch im chemischen Sinne schwach ist. „Es kann auch bedeuten, dass diese beiden Proteine einfach nur in einem sehr kleinen Teil der Zelle überhaupt aufeinandertreffen“, so Hein.
Denken wir an das leistungsfähige Quartett aus unserem Eingangsbeispiel: Jörg, Bernd, Hanna und Beate treffen nur einmal pro Woche zusammen, entwickeln dann aber jedes Mal Ideen, die die Firma voranbringen. Eine grafische Darstellung des sozialen Firmennetzwerks würde auf eine schwache Interaktion schließen lassen und Jörgs Zigarettenpartnerin Mia womöglich eine viel größere Bedeutung zuschreiben. Ebenso kann es passieren, dass man ein Protein unterschätzt, das vielleicht in einem einzigen Schritt der Mitose eine Schlüsselrolle spielt und dann nur ganz kurz mit dem Spindelapparat interagiert. Auch eine im biophysikalischen Sinne schwache Bindung könne zudem wichtige Signale vermitteln, ergänzt Hein.
Ob solch ein Interaktom aus HeLa-Zellen nun repräsentativ ist für alle menschlichen Zellen, mag man als kritische Frage einwerfen. „Es wird definitiv Unterschiede zwischen verschiedenen Geweben geben“, meint Hein hierzu. „Denn sonst hätten wir nicht so viele Zelltypen.“ Hein glaubt aber auch, dass es einen konstanten Kern des Netzwerks gibt, der in allen Säugerzellen ähnlich aussieht. „Man weiß bereits, dass viele Proteine in allen Geweben gleichermaßen zu finden sind – somit spricht vieles für einen universellen Teil des Interaktoms“, ist er sicher.
Womöglich kann dieses Interaktom künftig ja auch als Referenz für weitere Experimente aus der Proteomik dienen. Und ganz sicher wird das Netzwerk dann durch neue Daten aktualisiert und bleibt bis auf weiteres Work in Progress – wie derzeit alles in der Interaktomik.
Last Changed: 02.07.2018