Strukturprobleme
Quantifizierung von SH-Gruppen
Hubert Rehm
Eine der traumatischen Erfahrungen meiner Postdok-Zeit war der Versuch, die molekulare Struktur des synaptischen Vesikelproteins Synaptophysin zu ermitteln.
Mein Freund Bertram hatte das Protein zufällig über die Produktion von monoklonalen Antikörpern entdeckt. Es stellte sich heraus, daß Synaptophysin massenhaft in fast allen Neuronen und neuroendocrinen Zellen vorkam. Der Neurotransmitter spielte dabei keine Rolle. Weil Bertram Mediziner war und dauernd Klinikdienst schieben mußte, konnte er die Entdeckung nicht ausschlachten. Ich sollte ihm helfen und selbstlos tat ich das.
Synaptophysin, das stellte sich dank SDS-Gelelektrophorese schnell heraus, besaß ein Molekulargewicht von 38 Kd. Es war glykosiliert und ein integrales Membranprotein, denn es konnte nur mit Detergenzien in Lösung gebracht werden. Das scheinbar problemlos: 0,1% Triton X-100 reichten aus.
Was tat Synaptophysin in der Membran?
Vor der Funktion standen Bertram und ich wie ein Gespann Ochsen vorm neuen Scheunentor – eine Position, die die Wissenschaft heute noch einnimmt. Jedoch, wenn man ein Tor nicht öffnen kann, empfiehlt es sich, dessen Aufbau zu studieren. Bei Synaptophysin hieß das: Strukturbestimmung. Mit Strukturbestimmungen glaubte ich mich auszukennen, das hatte ich schon in meiner Doktorarbeit getan, das sollte ein schnelles Paper abwerfen und meine Aussichten auf eine Gruppenleiterstelle mehren. Damals hing ich noch dem fatalen Irrglauben an, es käme für den Aufstieg in der Wissenschaft auf Ergebnisse an.
Rätselhafte Struktur
Die erste Frage war: Aus welchen Untereinheiten besteht natives Synaptophysin? Reinigungsversuche von in Triton oder anderen nicht denaturierenden Seifen gelöstem Synaptophysin mit Ionenaustauscher- oder Immunoaffinitätssäulen ergaben nur das 38 Kd Protein. Also war solubilisiertes Synapotophysin mit keinem anderen Protein assoziiert, höchstens mit sich selber (Homopolymer). Das führte zur zweiten Frage, zur Merigkeit: Wieviele 38 Kd Untereinheiten enthielt der native Synaptophysinkomplex?
Die Zahl der Untereinheiten eines Proteins läßt sich mit (mindestens) drei Methoden bestimmen: Röntgenstrukturanalyse, Quervernetzung und Molekulargewichtsbestimmung. Die Röntgenstrukturanalyse fiel aus, denn einen Synaptophysin Kristall besaßen wir nicht und wußten auch nicht wie ein solcher herzustellen wäre. Blieben Quervernetzung und Molekulargewichtsbestimmung des Komplexes. Letztere schien uns das Einfachste, denn teilt man das Molekulargewicht des Komplexes (x Kd) durch das des Monomers (38 Kd) erhält man die Anzahl von Monomeren. Simple Rechnung aber die Praxis..! Zur Molekulargewichtsbestimmung des Komplexes, solubilisiert man das Protein in einem milden Detergens und unterwirft es einer Gelfiltration. Die ergibt den Stokes Radius des Komplexes. Darauf folgen Sucrosedichtezentrifugationen in H
2O und D
2O und deren Parameter ergeben zusammen mit dem Stokesradius das Molekulargewicht. Die Methode wurde schon 1975 von Steven Clarke angegeben und die Details finden Sie im Experimentator Proteinbiochemie (5. Auflage) auf Seite 122-123. Für den Synaptophysinkomplex erhielt ich ein Molekulargewicht von 119 Kd. Leider war die Rechnung zwar mathematisch exakt, physikalisch aber zweifelhaft: So muß z.B. das partielle spezifische Volumen des Proteinanteils des Komplexes angenommen werden. Zudem war nicht klar, ob der Zuckeranteil des Synaptophysins beim Molekulargewicht des Monomers mitzurechnen ist oder nicht. Im ersten Fall hätte man mit 38 Kd rechnen müssen im zweiten mit 34 Kd. Im ersten Fall hätte man ein Trimer angenommen im zweiten eher ein Tetramer. Zudem mußte die Synaptophysinstruktur in Lösung keineswegs die Verhältnisse in der Membran wiedergeben.
Vielleicht lag Synaptophysin in der Membran als Oktamer vor und zerfiel in wäßriger Lösung in zwei Teile? Oder umgekehrt: Es lag in der Membran als Dimer vor, das in Lösung – Gott weiß warum – zum Tetramer assoziierte. Dies war mir schmerzlich bewußt und daher versuchte ich die Ergebnisse über Quervernetzen mit Glutaraldehyd (vernetzt Aminogruppen) bzw. Cu
2+ o-Phenantroline (vernetzt -SH Gruppen) abzusichern. Mit membrangebundenem Synaptophysin erhielt ich mit allen Quervernetzern ein Produkt vom Molekulargewicht 76 Kd, vermutlich das Dimer. Gelöstes Synaptophysin jedoch verhielt sich anders: Schon beim Stehenlassen bei 4 °C bildeten sich außer dem Dimer noch Tri- und Tetramere. Zugabe von Cu
2+ o-Phenantroline gab das gleiche Ergebnis. Anscheinend bildeten sich zwischen den Homomeren Disulfidbrücken aus. In der Tat ergab die spätere Sequenzierung des Synaptophysins (Leube et al., 1987) vier Cysteinreste, die in jedem der zwei intravesikulären Loops des Proteins eine Disulfidbrücke bilden.
Konnte man also schließen, daß Synaptophysin ein Tetramer war?
Vielleicht. Auf dem Blot war zwar oberhalb vom Tetramer keine Bande mehr zu sehen aber das konnte daran liegen, daß diese Bande in dem leichten Schmier, der sich vom Auftrag herunterzog, untergegangen war. Vielleicht wurde sie auch wegen des hohen Molekulargewichts schlecht vom 8% Gel auf den Blot übertragen, oder war unlöslich und fiel aus ... Und warum sah man bei Quervernetzen von Vesikeln, also membranständigem Synaptophysin, nur Dimere?
Bertram und ich waren mit unserer Weisheit, unseren Methoden und unseren Nerven am Ende. Die Struktur des Synaptophysins schien ein ebenso zäher Brocken zu sein wie seine Funktion. Wir nannten es nur noch Syph. Wie Johnston & Südhof, 1990 später gezeigt haben, lag die Ursache unserer Probleme in den instabilen Disulfidbindungen. Die Struktur hätte sich zweifelsfrei nur unter Bedingungen bestimmen lassen, unter denen die Disulfidbindungen auch in Lösung stabil blieben. Um diese Bedingungen zu finden, hätte es eine Methode gebraucht, die die Anzahl der freien SH-Gruppen und Disulfidbindungen in membrangebundenem und gelöstem Syph angibt.
Dänische SH-Gruppen Bestimmung
Neulich stieß ich in Analytical Biochemistry auf ein Paper mit dem Titel "Quantification of protein thiols and dithiols in the picomolar range using sodium borohydride and 4,4'-dithio-dipyridine". Es war die Nummer 15 unter den "heißesten" Artikeln von Analytical Biochemistry und erschien online am 8. Januar 2007 – über 20 Jahre nach unseren Arbeiten zur Syphstruktur. Die Autoren, durchweg Dänen, erwähnen das Reagenz 5',5'-Dithiobis-(2-nitrobenzoesäure) DTNB, das schon 1959 von George Ellman eingeführt worden war. Das farblose DTNB reagiert mit Thiolat-Anionen in einer Thiol-Disulfid Austauschreaktion wobei eine Disulfidbindung entsteht und die gelbe 2-Nitro-5-Thiobenzoesäure (NTB) freigesetzt wird. Über die NTB Bildung läßt sich somit die Menge der SH-Gruppen bestimmen. Dieser Test war uns damals unbekannt. Eine Schande und ein Glück, denn er hat Nachteile: Die Reaktion läuft nur bei alkalischem pH ab, sie benötigt hohe Konzentrationen an gereinigtem Protein, DTNB ist instabil und die protonierte Form von NTB farblos.
Diese Nachteile, so die Dänen, könnten mit dem Thiolreagenz 4,4'-Dithiodipyridin (4-DPS) und Na-Borhydrid vermieden werden. 4-DPS reagiert ähnlich wie DTNB, jedoch auch im sauren Milieu und ist kleiner und hydrophober als DTNB. Es soll daher im hydrophoben Inneren der Proteine zuverlässiger reagieren. Als Nachweisreagenz entsteht das 4-Thiopyridon (Absorption bei 324 nm). Da 4-Thiopyridon säurestabil ist kann es auf der HPLC nachgewiesen werden. Zudem ist sein Extinktionskoeeffizient 50% höher als der von NTB. Die Nachweisgrenze des 4-DPS Tests liegt im Picomolaren also mehrhundertfach niedriger als der DTNB Test.
Mit beiden Tests lassen sich sowohl die freien SH-Gruppen als auch die Gesamtzahl der Cysteinreste eine Proteins bestimmen. Für Letzteres müssen die Disulfidbindungen vollständig reduziert werden. Danach muß das reduzierende Agens aus der Reaktionsmischung verschwinden, dann erst kann das Nachweisreagenz, also z.B. 4-DPS, zugegeben werden. Wer die üblichen Reduzierungsreagenzien wie Dithiothreitol oder Mercaptoethanol verwendet, muß hinterher einen wirksamen Reinigungsschritt anschließen: TCA-Fällung mit mehrfachem Waschen reicht nicht aus, Gelfiltration ist umständlich und führt zu Proteinverlusten und dies gilt auch für andere Säulen. Die Dänen lösen dieses Problem indem sie mit Na-Borhydrid reduzieren: Na-Borhydrid zersetzt sich vollständig im sauren Milieu, kann also durch Ansäuern entfernt werden.
Die Dänen haben ihre Methode mit Lysozym, BSA, RNase A, Carboxypeptidase Y ausprobiert. Die Proteine wurden mit 6 M Harnstoff denaturiert und mit Na-Borhydrid reduziert (30 min, 50 °C). Danach wurde HCl zugegeben und die Thiolgruppen mit 4-DPS bestimmt. Für Carboxypeptidase Y, die eine frei Thiolgruppe und fünf Disulfidgruppen enthält, erhielten die Dänen einen Wert von 11,4 Thiolgruppen. Auch bei den anderen Proteinen entsprachen die Messwerte den tatsächlichen Gegebenheiten.
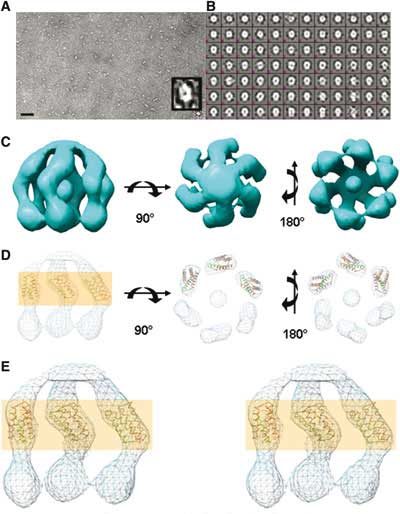
Wenn ich mir nicht geschworen hätte, nie mehr eine Pipette in die Hand zu nehmen, würde ich das Problem der Merigkeit von Syph (und das anderer Vesikelproteine) mit dieser Methode noch einmal angehen. Denn die Syphstruktur ist so unklar geblieben wie seine Funktion (Rolle bei der Vesikelbildung? Kationen Kanal?). Syph Knockout Mäuse sind scheinbar normal. Was die Struktur betrifft, so hatten wir uns 1986 – Oh, jugendlicher Leichtsinn! – auf ein Tetramer festgelegt (Rehm et al., 1986). Schon 1988 erschien ein Paper (auf dem ich auch noch Coautor war!) das eine hexamere Struktur in die Welt trompetete (Thomas et al.). 1990 nahm sich Patricia Johnston der Frage noch einmal gründlich an und kam zu dem Schluß, daß Syph wohl doch eher ein Tetramer als ein Hexamer sei. Danach schwieg der Blätterwald. Erst im Juni dieses Jahres legte Christopher Arthur, Colorado, in Structure (15, 707-714) nach: Mittels Elektronenmikroskopie habe er ein Hexamer gefunden. So fest wie die Felsen von Colorado scheint mir der Befund aber nicht zu stehen. Obwohl Arthur in Triton gelöstes Syph untersuchte, ignoriert er die Disulfidproblematik: Johnstons Arbeit scheint er nicht zu kennen, zumindest zitiert er sie nicht. Auch ist Arthur anscheinend unbekannt, daß man mit einer Gelfiltration Stokes radii bestimmt und keine Molekulargewichte. Nichts gegen Arthur – er muß ein netter Kerl sein, denn er hat mich zitiert – doch das letzte Wort zur Syphstruktur ist noch nicht gesprochen.
Welcher Doktorand auch immer den Ehrgeiz hat, dieses Wort zu sprechen, Bertram und ich wünschen ihm Glück. Er wird es nötig haben.
Letzte Änderungen: 23.10.2007