Enhancer-Suche leicht gemacht
Genomweit nach Enhancern zu suchen kostet Zeit und Geld. Die Gruppe um Alexander Stark am IMP Wien hat eine Methode entwickelt, die das Ganze beschleunigt.
(20. März 2013) Die Funktionen der nicht-kodierenden DNA im Zellkern sind komplex und noch weitgehend unerforscht, zumal die verwendeten Methoden relativ umständlich und zeitraubend sind. Ein Doktorand verbrachte früher oft mehrere Jahre damit, einzelne transkriptionsverstärkende Elemente für ein Gen (die sogenannten Enhancer) in der umgebenden genomischen DNA zu lokalisieren und mittels Reportergen-Assays zu charakterisieren. Die Gruppe um Alexander Stark vom Forschungsinstitut für Molekulare Pathologie (IMP) in Wien hat vor, die Kartierung und Funktionstestung von Enhancern mittels massiv-paralleler Sequenzierung zu beschleunigen. Dazu entwickelten sie eine neue Technik, die sie STARR-seq tauften (Science, 339(6123):1074-7).
Zählen durch Sequenzieren
„Der Clou von STARR-seq“, fasst Stark zusammen, „ist es, willkürlich aus einer genomweiten Bibliothek erzeugte Fragmente mit einer transkribierten Sequenz in einem Plasmid-Vektor zu koppeln – daher auch der Name self-transcribing active regulatory region sequencing, kurz STARR-seq.“ Das zu testende DNA-Fragment reguliert dabei die Produktion eines Transkripts, in dem es selbst enthalten ist. Das erlaubt die Lokalisation in der bekannten Genomsequenz und gleichzeitig eine Bestimmung der Aktivität.
Hierfür zerlegen die Molekulargenetiker um Stark zunächst die genomische DNA von Drosophila melanogaster mittels Ultraschall in Fragmente passender Größe von rund 600 bp. Diese klonieren sie in einen speziell konstruierten Reportergen-Vektor downstream eines minimalen Promotors und einer Protein-kodierenden Region (GFP). Es entsteht eine komplexe Bibliothek von Millionen Fragmenten, die 96 Prozent des (nicht-repetitiven) Genoms mindestens zehnfach abdecken. Den gesamten Pool an Plasmid-Klonen vervielfältigen sie und verwenden ihn zur Transfektion von Drosophila-Schneider 2 (S2)-Zellen. Nach 24 Stunden können sie die mRNA isolieren und die Reporter-spezifische RNA zu cDNA umschreiben.
„Die interessierenden Regionen am 3‘-Ende der RNA-Transkripte“, so Erstautor Cosmas Arnold, „die ja die potentiellen Enhancer-Kandidaten enthalten, amplifizieren wir weiter und sequenzieren sie schließlich unter Nutzung der initial ligierten Sequenzieradapter auf einer geeigneten Plattform – in diesem Fall Illumina/Solexa.“ Die erhaltenen paired-end reads von 2 mal 36 Basen Länge, deren Positionen im Genom exakt bestimmt werden können, geben die jeweiligen DNA-Fragmente an.

Die Anzahl der jeweiligen Fragmente im Sequenz-Pool der transfizierten Zellen wird schließlich ins Verhältnis zu ihrer Anzahl in der ursprünglichen Ausgangs-DNA-Bibliothek gesetzt und damit ein Maß für ihre regulierende Aktivität auf die Transkription des Reportergens ermittelt. „Die Auswertung der umfangreichen Sequenzierdaten kann innerhalb weniger Tage abgeschlossen werden“, bekräftigt Stark. „Mit den Ergebnissen kann man chromosomale Scans erstellen, in denen die transkriptionelle Aktivität von überlappenden DNA-Fragmenten entlang der Sequenzachse aufgetragen wird.“
Transkriptions-Netzwerke
Mit dieser Methode kartierten Stark und sein Team in S2-Zellen insgesamt 5.499 Elemente mit Enhancer-Aktivität. In den parallel getesteten ovarialen somatischen Zellen (OSC) waren es 4.682, die teilweise überlappten. Die Ergebnisse für die Enhancer, die starke Aktivitätsunterschiede zwischen den beiden Zelltypen zeigten, korrelierten gut mit der differentiellen RNA-Expression der flankierenden Gene. „Die getesteten Elemente funktionierten, wie man es von einem klassischen Enhancer erwartet, unabhängig von Position und Orientierung und weitgehend auch unabhängig vom verwendeten minimalen Promoter“, so Arnold. Die meisten Gene wurden von zwei bis drei Enhancern kontrolliert, es gab aber auch Gene mit bis zu zehn Enhancern in einem definierten Fenster um den Transkriptionsstart herum. Die stärksten Enhancer fanden die Wiener in Genen für Housekeeping-Proteine und für Transkriptionsfaktoren, eine Gruppe mit auffallend schwachen Enhancer-Aktivitäten waren dagegen die Gene für ribosomale Proteine. Die Mehrheit der identifizierten Enhancer war in Introns (besonders dem ersten) oder in intergenischen Regionen lokalisiert.
Natürlich muss eine Methode, die mit „nackter“ transfizierter DNA arbeitet, gegen etablierte Techniken wie dem Mapping von DNase I-hypersensitiven Stellen (DHS) oder Chromatin-IP (ChIP) bestehen, die auf Chromatin-Zugänglichkeit und Histon-Modifikation des endogenen Elements testen. Diese Validierung lieferten Stark et al. in ihrem Artikel in aller Ausführlichkeit mit.
Offene und verschlossene Enhancer
„Interessanterweise“, führt Stark aus, „ergab der Vergleich von STARR-seq mit DNase-hypersensitiven Stellen in den getesteten S2-Zellen, dass immerhin fast ein Drittel aller Enhancer mit starker Aktivität nicht im endogenen Chromatin-Kontext aktiv waren.“ Diese Enhancer werden anscheinend durch aktive Mechanismen supprimiert, wobei Histon-Acetylierung und -Methylierung eine Rolle spielen könnten. Diese Diskrepanz blieb auch erhalten, wenn diese „verschlossenen“ Enhancer nach stabiler Transfektion in die Chromosomen der S2-Zellen inkorporiert wurden. Auf der anderen Seite gibt es auch DHS-positive, sogenannte „offene“ Enhancer, die im STARRseq-Assay korrekt als inaktiv klassifiziert werden und wahrscheinlich Insulator-Proteine binden, also andere Funktionen haben.
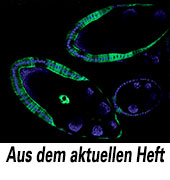
Da die Methode STARR-seq eine ganze Fülle weiterer Ergebnisse lieferte, die das Heft sprengen würden, sei hier nur noch ein Highlight herausgegriffen, dass gleichzeitig demonstriert, wie man in vivo-Relevanz mit den Möglichkeiten der modernen Genomforschung zeigen kann. Das Team um Stark brachte ausgewählte DNA-Fragmente, die in OSC-Zellen Enhancer-Aktivität zeigten, an das GAL4-System gekoppelt in definierte Genomregionen von Drosophila ein. Die so entstandenen transgenen Linien wurden mit Taufliegen aus der vorhandenen UAS-CD8-GFP-Bibliothek gekreuzt. „In den Ovarien der adulten Tiere aktivierten immerhin 11 von 13 getesteten Enhancern wie vorhergesagt die Expression des GFP und zeigten grüne Fluoreszenz, während die fünf negativen Kontrollen keine Expression zeigten“, so Stark.
Patente Methode
„Die Technik ist grundsätzlich auf beliebige Genome anwendbar“, betont Stark, der die STARRseq-Methode zusammen mit seinem team mittlerweile zum Patent angemeldet hat. Voraussetzung sei nur die Fähigkeit, eine repräsentative und ausreichend komplexe genomische DNA-Bibliothek zu erzeugen sowie der Zugang zu einer Hochdurchsatz-Sequenziereinrichtung. Als Proof of principle stellten die Wiener eine Bibliothek aus sechs humanen BAC-Klonen her, die in HeLa-Zellen erfolgreich getestet wurde. Hier wird es interessant sein, die Ergebnisse mit denen des multinationalen ENCODE-Projekts abzugleichen, die schon 300.000 Enhancer im menschlichen Genom vorhergesagt haben.
Fazit: Die IMP-Gruppe um Stark hat ein mächtiges Werkzeug geschaffen, um unter Nutzung moderner Sequenziertechniken genomweit Enhancer-Aktivitäten zu messen und dadurch Enhancer-Elemente zu kartieren. Gemeinsam mit komplementären Techniken wie DHS und ChIP ermöglicht STARR-seq die Erstellung von kompletten Genregulations-Netzwerken und könnte dadurch etwa in der Entwicklungsbiologie und Krebsforschung von Nutzen sein.
Thomas Schmitt
(Der Artikel erschien bereits im aktuellen Laborjournal 3-2013: 36-7; Fotos: Alexander Stark)
Letzte Änderungen: 19.04.2013