Tipp 218: Pull-Down-Assays mit Spike-In-Phosphopeptiden
Die Massenspektrometrie-basierte Phosphoproteomik identifiziert immer mehr Phosphoproteine, liefert jedoch keine Informationen zu deren Interaktionspartnern. Verwendet man die in der Massenspektrometrie häufig eingesetzten Spike-in-Phosphopeptide für Pull-Down-Assays, so kann man auch herausfinden, mit wem die Phosphoproteine wechselwirken.
Um ihr Überleben zu sichern, müssen Zellen reaktionsschnell und anpassungsfähig sein. Versucht ein pathogener Organismus in sie einzudringen oder sind die Wachstumsbedingungen perfekt, müssen die Zellen sofort reagieren und ihre umliegenden Nachbarn informieren. Wenn sie dazu erst ihre Transkription und Translation hochfahren müssen, dauert dies zu lange. Viel schneller und flexibler funktioniert die Antwort über Proteine, die durch Kinasen und Phosphatasen mithilfe von Phosphatresten posttranslational modifiziert werden. Proteomiker schätzen, dass dreißig Prozent aller Proteine eine oder mehrere Phosphorylierungen an ihren Serin-, Threonin- oder Tyrosin-Gruppen durchlaufen.
Schuss ins Blaue
Die Massenspektrometrie-basierte Shotgun-Proteomik, die meist für Proteom-Analysen verwendet wird, ist jedoch ein ziemlicher Schuss ins Blaue, und kaum für die Identifikation von Phosphoproteinen geeignet. Massenspektrometrische (MS)-Analysen verraten nur, ob und nicht wo die Modifikation auf einem Phosphoprotein sitzt. Schade, denn die Lage und die Abfolge der phosphorylierten Positionen sind oft ausschlaggebend für wichtige Eigenschaften, wie zum Beispiel Stabilität, Faltung, Interaktionen und subzelluläre Lokalisation.
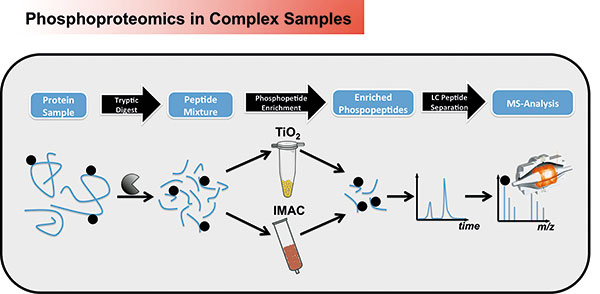
Inzwischen haben Proteomiker jedoch Techniken entwickelt, mit denen sie die Positionen und die Funktion von Phosphatgruppen in Proteinen bestimmen können. Der Weg zum Phosphoproteom führt zunächst über die Anreicherung phosphorylierter Proteine in einem Zellextrakt, zum Beispiel mit Titandioxid oder der Immobilisierten Metall-Affinitätschromatographie (IMAC). Die negativ geladenen Phosphoproteine werden mit diesen Techniken konzentriert und anschließend mit dem Massenspektrometer analysiert.
Die MS-basierte Phosphoproteomik produziert Unmengen von Daten, deren Verifizierung aber nur schleppend vorangeht. Ob zum Beispiel Protein X an Position Y konstitutiv oder stressabhängig phosphoryliert ist, wurde bislang vor allem mit spezifischen Antikörpern per Immunoblot nachgewiesen. Dies ist jedoch mit hohem Zeit- und Kostenaufwand sowie dem Risiko verbunden, dass der Antikörper das Antigen am Ende doch nicht erkennt.
Gezielte Massenspektrometrie
Ein eleganterer Weg führt über die gezielte Massenspektrometrie (Targeted MS). Bei dieser wird der Probe eine synthetische Variante des zu verifizierenden Phosphoproteins beigemengt und parallel analysiert (Targeted Spike-in Validation). Das synthetische Phosphopeptid muss dem endogenen Kandidaten möglichst genau entsprechen, um es im MS-Spektrum eindeutig zuordnen zu können. Außerdem lassen sich aus der bekannten Menge der eingesetzten Spike-in-Probe sowie mithilfe der jeweiligen Peak-Fläche im MS-Chromatogramm auch quantitative Aussagen treffen.
Die Sequenzen der Spike-in-Phosphopeptide sind identisch mit den Sequenzen ihrer endogenen Pendants. Sie werden jedoch mit Isotop-markierten Aminosäuren gespickt, in der Regel mit 13C- oder 15N-markiertem Lysin oder Arginin. Die Positionen der Markierungen passt man an die Schnittstellen der Protease Trypsin an, die für die enzymatische Fragmentierung vor der MS-Analyse eingesetzt wird.
Die Gruppe um Martina Schnölzer von der Functional Proteome Analysis-Einheit des Deutschen Krebsforschungszentrums in Heidelberg kam auf die smarte Idee, die Spike-in-Phosphopeptide nicht nur für die gezielte MS zu verwenden, sondern parallel dazu auch für Phosphorylierungs-abhängige Protein-Protein-Interaktionsstudien mit Pull-Down-Assays (Anal. Biochem. 568: 73-77).
Pull-Down mit Phosphopeptiden
Dazu koppelte sie die synthetischen Phosphopeptide zunächst an das Biotinylierungs-Reagenz NHS-LC-Biotin und entfernte nicht gebundenes NHS-LC-Biotin, das im weiteren Verlauf des Protokolls stören würde, mit der IMAC. Die biotinylierten Phosphopeptide immobilisierte das Team mithilfe von Streptavidin-beladenen (Magnet-)Kügelchen. Wie bei einem klassischen Pull-Down-Experiment sieht das Protokoll der Gruppe vor, dass man den Proteinextrakt einer Probe hinzufügt, nach dem Waschen verbliebenen Proteine eluiert, diese mit Trypsin verdaut und die erhaltenen Peptide anschließend massenspektrometrisch analysiert.
Das ist der grobe Plan, doch Schnölzers Team hat noch einige raffinierte Details in das Protokoll eingebaut, mit dem man zum Beispiel herausfinden kann, welche Interaktionspartner die phosphorylierte und welche die nicht-phosphorylierte Version des Peptids bevorzugen – beziehungsweise welche unabhängig von der Phosphorylierung binden.
Drei Phosphopeptid-Töpfe...
Hierfür benötigt man das synthetische Phosphopeptid im phosphorylierten und nicht-phosphorylierten Zustand, weshalb das Team einen Teil der biotinylierten Probe vor der Immobilisierung mit Phosphatase behandelte. Anschließend füllten die Heidelberger drei Gefäße mit den an die Streptavidin-Kügelchen gebundenen Phosphopeptiden. In zwei gaben sie die phosphorylierten Peptid-Varianten, in eines die dephosphorylierten.
Fehlten nur noch die Proteine, die in dem Pull-Down-Assay mit den immobilisierten Phosphopeptiden wechselwirken sollten. Um Proteine auseinanderhalten zu können, die von behandelten sowie unbehandelten Zellen stammten oder unspezifisch an die Phosphopeptide banden, fütterten die Heidelberger Zellkulturen mit Isotop-markierten Aminosäuren, die in die frisch synthetisierten Proteine der Zellen eingebaut wurden. Mit dieser Technik, die sich Stable Isotope Labeling by Amino Acids in Cell Culture oder kurz SILAC nennt, sind auch fein abgestufte Isotop-Markierungen möglich.
Schnölzers Mitarbeiter kultivierten die Zellen in Gegenwart von nicht-markierten, mittelschweren und schweren Aminosäuren. Die mit mittelschweren Aminosäuren aufgezogenen Zellen bestrahlten sie zusätzlich mit Röntgenlicht, um ihre DNA-Damage-Response (DDR) anzuregen.
...kombiniert mit drei Proben
Die Gruppe erhielt schließlich drei Zellkulturproben mit unterschiedlich stark Isotopen-markierten Proteinen: unbehandelt mit leichter Markierung, unbehandelt mit schwerer Markierung sowie Röntgen-bestrahlt mit mittelschwerer Markierung.
Schnölzer und Co. ließen der DDR nach der Bestrahlung zunächst zwei Stunden Zeit. Anschließend lysierten sie die Zellen und extrahierten die Proteine unter möglichst sanften Bedingungen, um ihre Konformationen oder mögliche Interaktionen nicht zu zerstören. Danach kombinierte das Team die Isotop-markierten Proteine mit den auf Magnetkügelchen immobilisierten Phosphopeptiden: Die leichten gaben die Forscher zu den dephosphorylierten Peptiden, die mittelschweren (behandelten) und schweren zu den phosphorylierten.
Nach einer Inkubationsphase und den obligatorischen Waschschritten eluierte die Gruppe die gebundenen Proteine mit einem SDS-PAGE-Probenpuffer und trennte sie in einem Gradienten-Gel. Die nach der Färbung erhaltenen Banden schnitten die Forscher aus, verdauten die darin enthaltenen Proteine mit Trypsin und analysierten die erhaltenen Peptide nach der Extraktion schließlich mit einem Orbi-Trap-Massenspektrometer.
Im MS-Chromatogramm pickten Schnölzers Mitarbeiter einzelne Peptide heraus und untersuchten das Verhältnis der Isotop-markierten Varianten. Peptide, deren Isotop-Varianten zu gleichen Anteilen auftraten, stammten offensichtlich von Proteinen, die unspezifisch an die Streptavidin-beschichteten Kügelchen gebunden hatten. Dominierten jedoch die mittelschweren und schweren Varianten, deutete dies auf eine Protein-Interaktion hin, die vom Grad der Phosphorylierung abhing. Trat nur die mittelschwere Variante gehäuft auf, so wies dies auf eine von der Phosphorylierung abhängige Interaktion mit einem stressinduzierten Protein hin.
Strategie funktioniert
Dass diese Strategie nicht nur in der Theorie funktioniert, demonstrierten die Heidelberger anhand der Phosphoproteine yH2AX und NUMA1 aus der humanen Zelllinie A549. Die Gruppe verifizierte mit ihrer Technik bekannte Interaktionspartner der beiden Proteine und spürte darüber hinaus auch neue auf. Zu Letzteren gehören viele Kandidaten, die (phosphorylierungs-)unspezifisch binden. Der eine oder andere davon könnte dennoch interessant sein. Um sie besser untersuchen zu können, müsste man das Zellkompartiment, in dem sie vorkommen, lediglich separieren, bevor man die Phosphoproteine mit der IMAC-Technik anreichert.
Andrea Pitzschke