Tipp 126:
Hausgemachte Mutagenese
Die Pfälzer Küche ist berühmt für ihren hausgemachten Saumagen. Mark Laible von der Uni Mainz und Kajohn Boonrod vom Neustadter Institut für Pflanzenforschung bevorzugen aber auch bei der zielgerichteten Mutagenese hausgemachtes.
Mutagenese Protokoll
Angesichts der Preise für Mutagenese-Kits sollte man sich genau überlegen, was man mutieren will. Wiederholungen wegen falscher Planung oder Pipettierfehlern sind nicht nur ärgerlich sondern auch teuer. Wer zudem wie wir mit dem Konstrukt seiner Wahl eine ganze Serie von Mutationen vor hat, merkt schnell, dass kommerzielle Mutagenese-Kits ziemlich ins Geld gehen.
Deshalb suchten wir im Internet nach einer Anleitung für eine "hausgemachte" Mutagenese. Dabei stießen wir auf verschiedene Protokolle, die sich nicht nur in Kleinigkeiten, sondern in grundsätzlichen Punkten widersprachen. So waren sich die Autoren zum Beispiel uneins, ob man phosphorylierte Primer braucht oder nicht, und ob man das mutierte Produkt vor der Transformation noch ligieren sollte.
Um uns in diesen Punkten Klarheit zu verschaffen, haben wir kurzerhand alle Vorschläge durchprobiert. Die
Pfu-Polymerase für die Amplifikation und das Restriktionsenzym
DpnI für den Verdau des Ausgangsplasmides kauften wir zu einem günstigen Preis. Zur Phosphorylierung der Primer benutzten wir T4-Polynukleotid-Kinase, für die Ligation T4-Ligase.
Wir testeten die folgenden vier Kombinationen: Phosphorylierte und unphosphorylierte Primer jeweils mit und ohne anschließende Ligation. Das Ergebnis war erstaunlich. Es stellte sich heraus, dass es im Prinzip völlig egal ist, ob man phosphorylierte Primer verwendet oder das Produkt nach der Mutagenese ligiert. Bei der einfachsten Variante mit unphosphorylierten Primern und ohne Ligation bekamen wir zwar am wenigsten Kolonien. Diese zeigten dafür die höchsten Mutationseffizienzen - elf aus zwölf gepickten Klonen trugen das erfolgreich mutierte Plasmid.
Seit diesem Tag verwenden wir nur noch dieses Protokoll und sparen dadurch eine Menge Geld. Eine Reaktion mit dem Kit, den wir bis dahin verwendet hatten, kostete etwa 25 Euro. Die Reaktion mit der hausgemachten Methode liegt bei knapp unter drei Euro. Selbst wenn man ein teureres Enzym wählt als wir, kostet die Reaktion immer noch nur knapp über drei Euro. In Prozent ausgedrückt ist das eine Kostenersparnis von über 400 %.
Fazit: Die zielgerichtete Mutagenese funktioniert auch ohne teuren Kit und mit normalen kompetenten Zellen. Phosphorylierte Primer und die
in vitro Ligation des mutierten Plasmids sind nicht nur umständlich sondern auch völlig unnötig.
So funktionierts
Für unsere hausgemachte zielgerichtete Mutagenese verwenden wir die von Papworth
et al. beschriebene Methode (Papworth, C., Bauer, J. C., Braman, J. and Wright, D. A. (1996)
Strategies 9(3):3-4.). Mit ihr kann man in Plasmiden Basenaustausche, Deletionen oder Insertionen von wenigen Nukleotiden einführen. Die Einsatzmöglichkeiten sind vielfältig und im Grunde nur durch die eigene Kreativität begrenzt. Die Methode funktioniert sehr gut mit Plasmiden bis zu einer Länge von 9 kb. Größere Plasmide sind schwieriger zu mutieren und man muss die Bedingungen etwas variieren. Für die Mutagenese benötigt man 10 bis 60 Nanogramm des zu mutierenden Ausgangsplasmids, das aus einem methylierenden (dam+)
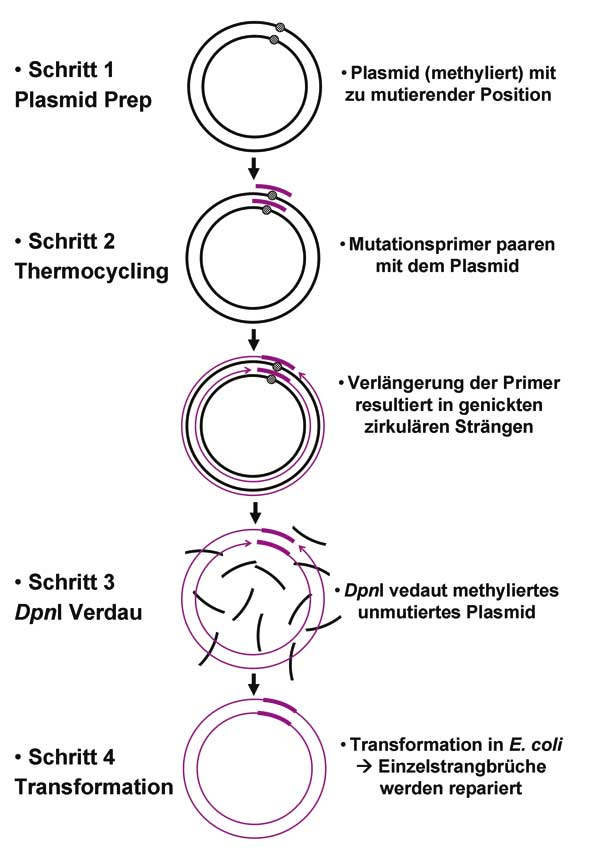
E. coli-Stamm stammen muss. Dazu kommen zwei komplementäre Primer mit Mismatches, die die gewünschte Mutation in das Plasmid einführen. Von jedem Primer setzt man ungefähr 150 Nanogramm ein (100 pM Stammlösung 1:10 verdünnen, davon 1,5µl entnehmen). Dazu braucht man noch eine thermostabile Polymerase, die Blunt-Ends generiert und eine Korrekturlesefunktion besitzt wie zum Beispiel die
Pfu-Polymerase. dNTP-Mix, Polymerase-Puffer und Wasser vervollständigen den Reaktionsansatz. Das komplette Plasmid wird im Thermocycler während eines Zyklus einmal repliziert, wobei die Mutationsprimer eingebaut werden. 18 Zyklen reichen aus, um genug mutiertes Plasmid zu generieren. Die niedrige Zyklenzahl spart zudem Zeit. Letzteres ist gar nicht so nebensächlich, denn bei einer Synthesegeschwindigkeit der
Pfu-Polymerase von 1 kb pro Minute und Denaturierungs- und Annealingphasen von jeweils 30 Sekunden bzw. einer Minute kann die PCR schon etwas dauern.
Nach beendeter PCR kontrolliert man die Reaktion auf einem Agarosegel. Mit fünf Mikrolitern sollte eine deutliche Bande zu sehen sein. Ist das der Fall, muss das Ausgangsplasmid aus dem Reaktionsansatz entfernt werden, um den Hintergrund an unmutiertem Plasmid gering zu halten. Hierzu werden zwei Mikroliter
DpnI zum Reaktionsansatz gegeben und für ein bis zwei Stunden bei 37 °C inkubiert. Da
DpnI nur methylierte DNA schneidet, baut sie nur das Ausgangsplasmid ab, während das frisch synthetisierte, mutierte Plasmid erhalten bleibt. Danach können kompetente Zellen der Wahl direkt mit fünf Mikrolitern aus dem Reaktionsansatz transformiert werden. Alle Schritte bis zur Transformation lassen sich trotz der langen Zeit im Thermocycler bequem an einem Tag durchführen.
Theorie und Praxis
Am nächsten Tag sollten laut Theorie nur Klone auf der Platte zu finden sein, die das mutierte Plasmid tragen. Dies ist jedoch nicht einmal bei Verwendung von teuren Kits der Fall. Man benötigt also eine Screening-Methode. Dazu führen wir mit der Mutation gleichzeitig eine Restriktionsschnittstelle ein oder deletieren eine entsprechende Schnittstelle. Ist keine vorhanden oder ist das gleichzeitige Einführen einer Schnittstelle mit der gewünschten Mutation nicht möglich, führt man erst eine Schnittstelle ein, und deletiert diese später mit dem Einführen der gewünschten Mutation. So dauert das Prozedere zwar doppelt so lange, doch hat man erst einmal einen Plasmid mit Markerschnittstelle, kann man diesen für die komplette Versuchsreihe verwenden.
Danach wählt man einige Kolonien aus und testet mit einem Restriktionsverdau ob die Markerschnittstelle vorhanden ist oder nicht. Wir picken immer sechs bis acht Klone pro Mutante von denen typischerweise 80 % die Mutation tragen. Blind verlassen sollte man sich allerdings nicht darauf und die Mutation durch Sequenzierung verifizieren. Wir hatten bisher jedoch noch keinen Fall, bei dem die Mutation im Verdau angezeigt wurde und dann bei der Sequenzierung fehlte
Design der Mutationsprimer
- Die Primer müssen komplementär zueinander und zwischen 25 und 45 Nukleotiden lang sein.
- Die gewünschten Mutationen müssen auf beiden Primern enthalten sein und sollten in deren Mitte liegen, beidseitig flankiert von mindestens 8 unmutierten Nukleotiden.
- Die Primer sollten einen GC-Gehalt von mindestens 40 % haben und 5' und 3' auf ein oder mehrere G oder C enden.
- Die Primer müssen nicht phosphoryliert und auch nicht FPLC- oder PAGE-aufgereinigt sein. Man sollte sie lediglich entsalzen.
- Zur Berechnung von Tm braucht man keine spezielle Formeln, sie sollte jedoch bei Primern von 25-30 Nukleotiden größer sein als 60 °C (Angaben der Hersteller). Als Annealingtemperatur ist 55 °C ein guter Richtwert. Sollte nach der Reaktion keine Bande auf dem Gel zu sehen sein, kann man die Annealingtemperatur variieren (50 °C oder 60 °C ausprobieren).
- Tipp: Für das Auffinden von Restriktionsschnittstellen, die man mit der Mutation einführt, eignet sich das Enzyme-Finder-Tool auf neb.com. Dort einfach den DNA-Code der gewünschten Aminosäuresequenz im Ambiguity Code eingeben.
Mark Laible(Uni Mainz) &
Kajoohn Boonrod (AlPlanta, Institut für Pflanzenforschung, Neustadt Weinstraße)
Letzte Änderungen: 17.10.2008