Next-Generation-Proteomik: Räumliche Proteomik: Mikroskopie trifft auf Massenspektrometrie
Im Gespräch mit Lisa Schweizer und Thierry Nordmann
Interview: Mario Rembold, Laborjournal 09/2022
(06.09.2022) Die Massenspektrometrie ist mittlerweile sensitiv genug, um Proteine einzelner Zellen zu erfassen. Mithilfe geeigneter Bildanalyse-Software lässt sich auch die räumliche Information speichern und später rekonstruieren.
Spezialisierte Zelltypen haben eigene proteomische Profile. Doch auch die zelluläre Nachbarschaft ist relevant, denn Signale im Gewebe werden zwischen den Zellen ausgetauscht. Die räumliche Information geht aber verloren, wenn man Zellen vor der Analyse in Suspension bringt. In der räumlichen Proteomik bezieht man diesen räumlichen Kontext mit ein.
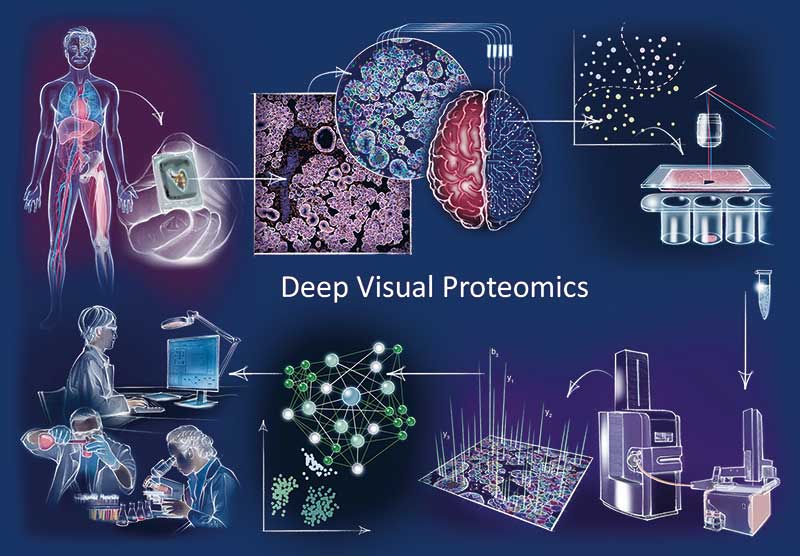
Am Max-Planck-Institut für Biochemie in Martinsried entwickelt die Abteilung Proteomik und Signaltransduktion von Matthias Mann Methoden für den Proteinnachweis und setzt vor allem auf die Massenspektrometrie. Im Frühjahr stellte das Team zusammen mit internationalen Kollaborationspartnern eine neue Methode vor: Deep Visual Proteomics. Für diese setzt es auch die Bildbearbeitungssoftware BIAS ein (Nat. Biotechnol. 40: 1231-40). Eine der Autorinnen ist Manns PhD Studentin Lisa Schweizer. Auch ihr Kollege Thierry Nordmann, ebenfalls aus der Mann-Gruppe, nutzt die räumliche Proteomik. Als Wissenschaftler und Facharzt für Dermatologie hofft er mit der Technik, seltene und insbesondere letale Hauterkrankungen künftig besser zu verstehen.
Schweizer und Nordmann haben mit uns über den aktuellen Stand der räumlichen Proteomik und die Ergebnisse im aktuellen Paper gesprochen.
Laborjournal: Um Proteine in einem fixierten Gewebe räumlich nachzuweisen, ist es doch am einfachsten, Antikörper zu verwenden. Dann sieht man direkt im Mikroskop, wo welches Protein sitzt. Andererseits kann man maximal 40 bis 50 Antikörper gleichzeitig verwenden. In diesem Punkt liegt dann wieder der Vorteil der Massenspektrometrie, denn auf diesem Weg lassen sich prinzipiell alle Proteine aufspüren. Trotzdem scheint mir zunächst die Methode mit leuchtenden Antikörpern eleganter, wenn ich wissen möchte, wo ein bestimmtes Protein vorkommt.
Lisa Schweizer » Das Schöne an der Massenspektrometrie ist, dass die Methode unabhängig von einer zuvor aufgestellten Hypothese sein kann. Wir können mittels Massenspektrometrie das Proteom anschauen, ohne vorher zu wissen, was wir suchen; wir müssen uns nicht auf ein vorher ausgewähltes Subset von Targets und Antikörpern beschränken. Mit Deep Visual Proteomics können wir diesen Teil aber dennoch integrieren und noch viel weiter darüber hinausgehen. Wir können beispielsweise mit Antikörpern Gewebe färben, wir können aber auch anhand von Merkmalen wie der Zellmorphologie Zellen auswählen, die danach extrahiert werden. Und darin lässt sich dann das vollständige Proteom analysieren.
Aber kann man im Massenspektrometer wirklich vollkommen neue Proteine entdecken? Ein Protein ist ja sehr viel komplexer als ein kleines Molekül – und selbst da kann es eine Herausforderung sein, Isomere mit gleicher Summenformel zu charakterisieren. Um ein Protein seinem „Fingerabdruck“ zuzuordnen, braucht man Referenzdaten. Jemand muss also bereits eine passende Signatur in einer Datenbank hinterlegt haben, das Protein war also schon bekannt.
Schweizer » Es gibt für Proteine FASTA-Files, also Referenzbibliotheken, die wir nutzen, nachdem wir in unserem Massenspektrometer die Proteine zerlegt haben – zunächst in Peptide, dann in Fragmente. Das können wir dann wie ein Puzzle wieder zusammensetzen. Das Schöne ist, dass beim Menschen das komplette Proteom identifiziert und verfügbar ist, also alle Proteine, zum Teil auch mit Isoformen. Was wir nicht so leicht finden können, sind zum Beispiel Splicing-Varianten. Aber auch dazu kann man natürlich optional eine angepasste Datenbank in die Auswertung einbeziehen. Das heißt, die einzige Grenze ist das bereits bekannte Proteom.
Vor wenigen Jahren war es noch eine große Herausforderung, Proteome in winzigen Probenmengen zu analysieren. Denn im Gegensatz zu Nukleinsäuren in der Transkriptomik kann man geringe Mengen von Proteinen nicht bis zu einer leicht nachweisbaren Menge amplifizieren. Ein Massenspektrometer muss also mit dem auskommen, was in der Originalprobe vorhanden ist. Inzwischen hat sich die Sensitivität so weit verbessert, dass man Proteomanalysen in einzelnen Zellen durchführen kann. Wodurch wurde dieser Fortschritt der vergangenen Jahre möglich?
Schweizer » Besonders wichtig war sicher die technologische Entwicklung an den massenspektrometrischen Geräten selbst. Dazu hat unser Labor Hand in Hand mit der Industrie zusammengearbeitet, um die Sensitivität zu erhöhen. Es gab also ein Update in der Hardware. Wichtig ist aber auch, was vor der Massenspektroskopie passiert. Da wurde die Flüssigkeitschromatografie optimiert; es gibt moderne Verfahren, um Moleküle schon vorher aufzutrennen und damit die Komplexität der Proben im Massenspektrometer herabzusetzen. Als dritten Punkt dieser Triade haben sich auch die bioinformatischen Methoden verbessert. Das Rückmappen der Fragmente gestalten wir heute viel effizienter, wir sind besser darin, die Komplexität der Spektren zu interpretieren.
Die Verbesserung der Sensitivität war eine wichtige Voraussetzung für die räumliche Proteomik, so wie Sie sie heute verstehen, oder?
Thierry Nordmann » Absolut. Wenn man bedenkt, dass wir für die Forschung auch Gewebeschnitte von Patienten analysieren möchten, die aus Biodatenbanken kommen und vielleicht schon zehn Jahre fixiert sind, haben wir natürlich keine Idealbedingungen mehr. Darin liegt dann die Herausforderung: Die Methode muss nicht nur mit HeLa-Zellen ohne räumliche Auflösung funktionieren, sondern auch mit echten Patientenproben und wir müssen diese Daten dann in eine räumliche Karte integrieren können. Dabei spielt die Sensitivität natürlich eine entscheidende Rolle.
Ich finde die molekulargenetischen Methoden elegant, bei denen man jedem Transkript im Probenmaterial einen eindeutigen Barcode „anheftet“. Darin kann auch eine räumliche Information stecken, wenn man zum Beispiel einen Gewebeschnitt auf eine Oberfläche klebt, auf der räumlich verteilt Primer mit verschiedenen Barcodes angebracht sind. Bei dieser Art räumlicher Transkriptomik muss man sich später keine Gedanken mehr machen um einzelne Proben. Alles kann in einem Tube weiterverarbeitet und sequenziert werden; der Barcode ist immer mit dabei. In der Proteomik geht das nicht. Stattdessen löst man Material aus der Probe heraus und muss diese Position dann für die spätere Auswertung speichern. Proben verschiedener Orte und Populationen dürfen nicht vertauscht oder gar gemischt werden. Für Ihre Methode der Deep Visual Proteomics verwenden Sie einen Laser, um Stücke herauszulösen. Wie exakt und präzise kann man mit dem Laser denn zielen und schneiden? Letztlich ist diese Auflösung während der Probennahme ein wesentlicher Flaschenhals, denn man möchte ja nur Zellen einer bestimmten Population herausschneiden. Vielleicht können Sie die Methode anschaulich beschreiben.
Nordmann » Im ersten Schritt haben wir das Gewebe. Derzeit arbeiten wir primär mit in Formalin fixiertem Gewebe, das in Paraffin eingebettet ist. Genauso, wie das auch in der klassischen Pathologie üblich ist, fertigen wir daraus 2,5 bis 3 Mikrometer dicke Schnitte an und legen sie auf Objektträger. Die können wir auch färben, zum Beispiel mit Hämatoxylin-Eosin oder, wie Sie es vorher erwähnt haben, auch mit verschiedenen Antikörpern, wenn wir bereits eine spezifische Zellpopulation im Sinn haben, die uns besonders interessiert.
Anschließend scannen wir diesen Schnitt auf einem ganz normalen Mikroskop. Jetzt haben wir die visuellen Daten dieses Schnittes. Die können wir als Matrix benutzen, um die Zellen, die uns interessieren, später herauszufiltern. Dafür haben wir die Software BIAS (Biology Image Analysis Software) entwickelt. Dieses Tool ermöglicht eine zielgenaue Segmentierung der Zellen nach Parametern, die man selbst bestimmen kann. Also zum Beispiel nach Zellgröße, Morphologie oder nach der Signalintensität eines Fluoreszenzfarbstoffs. Man kann die Software auch eigenständig eine Machine-Learning-Klassifikation benutzen lassen, um das Bild zu segmentieren. Daraufhin wird eine digitale Kontur erstellt für jede Zelle oder Struktur, die man möchte. Die wird, je nach Algorithmus, in verschiedene Klassen unterteilt und als XML-File ausgelesen. In solch einer Datei liegt dann die digitale Kontur des gesamten Slides in Einzelzellen vor.
Schweizer » Diese Schablone können wir dann elektronisch auf unser Laser-Capture-Microdissection-Mikroskop übertragen. Wir haben also einmal das physische Gewebe unter dem Mikroskop, aber computerbasiert projiziert das Mikroskop diese digitale Schablone auf den Schnitt. Um nun auf die Frage der Präzision zurückzukommen: Der Laser kann sowohl zelluläre als auch subzelluläre Strukturen ausschneiden – zum Beispiel die Zellkerne bei größeren Zellen wie Epithelzellen. Die meisten Zelltypen können wir auf Einzelzellebene ausschneiden.
Mit „Bildsegmentierung“ ist gemeint, in einer Bilddatei Regionen zu erkennen und voneinander abzugrenzen. Solche „Segmente“ können dann einzelne Zellen sein, aber die Grenzen lassen sich auch nach anderen Kriterien festlegen – vielleicht größere Zonen im Gewebe, in denen ein bestimmter Fluoreszenzmarker anschlägt und die Grafik einen anderen Farbwert hat. Oder eine subzelluläre Struktur wie der Zellkern.
Schweizer » Genau. An diesem Punkt bedienen wir uns dann der normalen Immunohistochemie und Immunofluoreszenz, zum Beispiel, um Zellmembranen oder Kerne zu färben und als Strukturen zu erkennen. Klassischerweise interessiert uns da die Zellmembran. Und optimalerweise auch noch die Lage des Kerns. So können wir dann Umrandungen von Zellen unterscheiden von Umrandungen, die nur einen leeren Raum einschließen. Und über geeignete Marker können wir auch Zelltypen auseinanderhalten, in denen etwas hoch- oder herunterreguliert ist.
Und wie kommt das ausgewählte Stück vom Objektträger in das Gefäß, das dann weiter für die Massenspektrometrie aufbereitet wird? Der Laser löst ja ein Stück heraus. Liegt der Objektträger mit der Probe dann umgekehrt in der Apparatur?
Schweizer » Richtig, die Probe liegt sozusagen verkehrt herum auf dem Mikroskop. Im Fokus-Punkt schneidet der Laser dann durch das Gewebe hindurch. Dabei fährt er wie ein Stift um diese Zellen herum und schneidet die Kontur nach, die der Computer vorgegeben hat. Zunächst ist das also bloß ein Schnitt. Dann gibt es einen zentralen Puls, der diese Shape herauslöst. Sie wird senkrecht darunter in einem Kollektor aufgesammelt. Wir sammeln das Material in 384-Well-Platten, sodass wir auf einer Platte also 384 Proben gleichzeitig prozessieren können.
Mit der Software BIAS haben Sie das Rad aber nicht neu erfunden, denn es gibt diverse Tools für die Analyse und Segmentierung biologischer Bilddaten. Im Paper schreiben Sie von einem „Benchmarking“, das Sie mit BIAS durchgeführt haben gegenüber zwei weiteren Tools zur Bildsegmentierung: unet4nuclei und Cellpose. Was genau ist das Besondere an BIAS?
Schweizer » Die Bildanalyse ist natürlich ein sehr weites Feld und keine Erfindung speziell für Proteomics. Was BIAS so besonders macht, ist die Arbeitsoberfläche, die von Anfang bis zum Ende darauf ausgelegt ist, das experimentelle Design von Deep Visual Proteomics zu verfolgen. Wir können aus verschiedenen Mikroskopen unsere Dateien laden und sehr leicht und zugänglich diese Analyse durchführen. Im grafischen Benutzer-Interface braucht man auch keine Programmierkenntnisse. Man kann diesen Workflow sehr einfach durcharbeiten bis hin zum Export der Information über jene Zellen, die man gerne ausschneiden will. Dabei sind auch verschiedene Mikroskop-Modelle berücksichtigt, inklusive den unterschiedlichen Arten, wie sie Proben sammeln. Also ob der Laser einmal spiralförmig um die Shapes herumgeht oder ob er den kürzesten Weg nimmt.
Ist die Software bereits für jeden verfügbar oder war das Paper zunächst nur ein Proof of Concept?
Nordmann » Die kann jeder benutzen es gibt dafür eine Lizenz zu kaufen. BIAS ist also vergleichbar mit anderen gängigen Segmentierungsmethoden, die bereits existieren und die wir auch mit in die Benchmark eingeschlossen haben. BIAS ist sehr einfach zu bedienen und funktioniert hervorragend. Das Tool ist auf diese Pipeline zugeschnitten, doch es kann noch viel mehr.
Was zum Beispiel?
Nordmann » Zum Beispiel könnten Sie ein Assay designen, um verschiedene Zellen zu quantifizieren. Da könnte eine Frage sein: Wie ist der prozentuale Anteil dieser Zellen im Vergleich zur Gesamtpopulation? Sie können diese Zellen dann entsprechend segmentieren und die Statistik in BIAS ausführen.
BIAS ist also gar nicht allein auf Proteomik limitiert?
Schweizer » Richtig. Theoretisch können Sie die Software für alle möglichen Anwendungen nutzen, denn es ist ein sehr modulares System. Aber wir haben BIAS als Interface zwischen einer hochauflösenden Bildgebung und hochsensitiver Massenspektrometrie und dem sehr präzisen Laser-Capture-Microdissection-Mikroskop optimiert.
In BIAS kommt für die Mustererkennung auch maschinelles Lernen zum Einsatz. Muss der Nutzer das Programm erst noch selbst trainieren oder gibt es bereits Bibliotheken, die man für bestimmte Strukturen verwenden kann?
Schweizer » Bis zu einem gewissen Grad kann man das Training sehr leicht zugänglich in der Software durchführen. Es gibt aber schon eine Standardbibliothek für verschiedene Zelltypen, die konstant ausgeweitet wird. Dazu kommen also regelmäßige Updates. Man kann aber innerhalb einer Probe auch selbst das Machine Learning verwenden, um die Präzision für Zellerkennung und Klassifizierung zu erhöhen. Denn jede Probe ist ja unterschiedlich.
Für das aktuelle Paper zur Deep-Visual-Proteomik haben Sie Eileitergewebe untersucht, um zu zeigen, dass die Methoden funktionieren. Warum ausgerechnet dieses Gewebe?
Schweizer » Das ist tatsächlich ein Experiment, das ich selbst mit den Erstautoren der Publikation zusammen in Kopenhagen durchgeführt habe. Die Wahl fiel auf dieses Gewebe, weil man daran die Unterscheidung zwischen zwei verschiedenen Zelltypen exemplarisch sehr schön sieht. Dort gibt es einmal Zellen mit Zilien und die sekretorischen Zellen ohne Zilien auf der Oberfläche. Anhand verschiedener Marker kann man diese sehr klar voneinander differenzieren. Wir haben in der Publikation FOXJ1 genutzt, da die Zellen mit Zilien FOXJ1-positiv sind, die sekretorischen Zellen hingegen nicht. Man kann die Bilder außerdem morphologisch auswerten, da man das Zilium in dieser Färbung sehr gut sieht.
Mit dieser Strategie konnten wir nach der Massenspektrometrie sehen, dass der Marker FOXJ1 nur in der Zellpopulation mit Zilien nachweisbar ist. In allen anderen Zellpopulationen tauchte er nicht auf. Und das, obwohl diese Zellen im Eileiter direkt nebeneinanderliegen. Somit konnten wir hier zeigen, dass der Laser diese Zellen sehr genau extrahieren kann und die Software präzise zwischen beiden Zelltypen unterscheidet.
Und es waren um die 5.000 unterschiedliche Proteine, die sie pro gemessener Zellpopulation massenspektrometrisch nachweisen konnten.
Schweizer » Ja. Die Anzahl der Proteine ist natürlich gewebespezifisch. Schön war, dass unser Marker ein Transkriptionsfaktor ist, und die sind ja klassischerweise eher niedrig abundant in der Zelle. Wir sehen also nicht nur die hoch abundanten Proteine, sondern auch die, die in sehr geringer Anzahl vorkommen.
Können Sie auch die Proteinmengen bestimmen?
Schweizer » Man kann die Proteinprofile miteinander vergleichen. Wichtig ist, dass die Proben dann in einer Platte gemeinsam prozessiert werden, um Abweichungen zu vermeiden. Die Leistung unseres Massenspektrometers ist sehr robust und reproduzierbar. So kann man die Fingerprints verschiedener Zelltypen miteinander vergleichen. In unserem Experiment haben wir nicht nur bereits bekannte Marker für Zellen mit und ohne Zilien gefunden, sondern wir konnten auch neue potenzielle Marker identifizieren, die mit den bekannten Markerproteinen korrelieren und ebenso nur in einem der beiden Zelltypen vorhanden sind. Und wir können feststellen, ob gemeinsam vorkommende Proteine in einer Zellpopulation höher oder niedriger abundant sind.
Lassen sich so auch absolute molare Mengen der Proteine ermitteln? Oder sind das immer relative Vergleiche?
Schweizer » In der Massenspektrometrie arbeiten wir eher mit relativen Vergleichen. Es gibt Methoden, mit denen man absolute Werte bestimmen kann. Zum Beispiel gibt es den sogenannten Proteomic Ruler, um Proteinmengen recht genau zu bestimmen. Oft geht es uns aber tatsächlich um die differenzielle Regulation. Das heißt, wir vergleichen eine Probe mit einem Kontrollgewebe oder wir vergleichen zwischen verschiedenen Zellpopulationen.
Welche Herausforderungen sehen Sie für die Zukunft, wenn es speziell um räumliche visuelle Proteomik geht?
Nordmann » Mein größtes Ziel als Kliniker ist, dass man diese Methode für klinisch relevante Fragestellungen einsetzt und Patienten direkt von ihr profitieren. Primär verwenden würde ich sie entsprechend, um Krankheiten zu erforschen, die selten sind und für die keine etablierten Therapiestrategien existieren. Da wäre eine Herausforderung, über Deep Visual Proteomics Krankheitsmechanismen besser zu verstehen und daraus neue Therapieformen abzuleiten.
Schweizer » Da kann ich nur zustimmen: Es gibt viele Herausforderungen, aber auch Hoffnungen. Wir wollen dieses Feld revolutionieren und diese Revolution auch umsetzen – also die Kollegen davon überzeugen, dass die Extraktion einzelner Zelltypen aus dem Gewebe einen enormen Vorteil für die proteomische Analyse hat.
Zum Beispiel um Krebsmetastasen zu verstehen, die aus einem anderen Gewebe in ein Zielgewebe gewandert sind. Die können wir jetzt aus dem Hintergrund dieses Zielgewebes extrahieren, anstatt nur einen Mittelwert der proteomischen Signaturen zu bestimmen. Auch die Heterogenität in einem Tumor können wir nur über die Einzelzell-Proteomik erfassen, denn im Bulk-Ansatz geht diese Information verloren.
Läuft dazu ein konkretes Projekt bei Ihnen?
Nordmann » Derzeit ist noch nichts veröffentlicht, aber ich schaue mir entzündliche Hauterkrankungen auf Proteinebene an und will diese zusammen mit der räumlichen Komponente verstehen. Seit meinem Studium interessiert mich dabei die immunologische Seite. Deswegen analysiere ich eine Vielzahl inflammatorischer Dermatosen, um zu verstehen, welche Immunzellen in welcher Relation zu den Keratinozyten stehen. Welche Moleküle sezernieren sie? Was ist ihr Profil? Wie richten sie Schäden an im Vergleich zu gesunder Haut? All das mit dem Ziel, Therapiestrategien zu entwickeln.
Nun könnte man doch genau solche Zelltypen wie Leukozyten ziemlich leicht über ein FACS von den Keratinozyten separieren und sammeln. Denn auch bei Ihrer Methode geht es Ihnen ja nicht um die einzelne Zelle, sondern letztlich um Zellpopulationen. Wäre der Weg über eine Zellsortierung in diesen Fällen nicht viel einfacher? Oder ist die räumliche Information hier wirklich so wichtig?
Nordmann » Mit unserer Methode haben wir zunächst einmal den Vorteil, dass wir auch in Biobanken und Archiven Proben suchen können. Das ist enorm wertvoll gerade für die Erforschung seltener Erkrankungen, denn mit einer durchflusszytometrischen Analyse könnten wir hauptsächlich prospektive Studien machen und bräuchten erstmal Jahre, um ausreichend Material dieser seltenen Zellen zu bekommen.
Weil man das archivierte formalinfixierte Gewebe nicht für einen Zellsortierer aufbereiten kann?
Nordmann » Genau. Und ich spreche wirklich von Proben, die zwanzig Jahre oder älter sein können. Der zweite große Vorteil: In einer durchflusszytometrischen Analyse bringen Sie die Zellen in Suspension und verlieren jede räumliche Information. Die Biologie definiert sich aber auch über die Räumlichkeit. Zellen verhalten sich anders, wenn sie in einem anderen Milieu sind. Und genau diese Feinheiten können wir mit der räumlichen Proteomik untersuchen.