Es kommt auf jede einzelne Zelle an - Einzelzell-Transkriptomik
Mario Rembold
(10.03.2022) Ein Barcode für jede Zelle – auf diesen einfachen Nenner könnte man moderne Methoden der Einzelzell-Transkriptomik bringen. Es gibt aber noch einiges zu optimieren. Und auch die räumliche Rekonstruktion der Transkripte ist nicht ganz einfach.
Die mRNA eines Organismus verrät, welche Gene gerade ausgelesen werden. Organe oder Gewebe bestehen aber aus vielen Zellen mit verschiedenen Aufgaben, und auch Tumorzellen sind mitunter sehr heterogen. Um wirklich zu verstehen, wann welche Gene ein- oder ausgeschaltet werden, muss man einzelne Zellen untersuchen.
Was vor einem Jahrzehnt noch eine Sensation war, ist inzwischen keine Hexerei mehr. Das Sequenzieren vieler einzelner RNA-Moleküle im Hochdurchsatz ist heute Standard. Viel kniffliger ist die Aufgabe, die vielen Sequenzdaten individuellen Zellen zuzuordnen. Hierfür hängt man der cDNA eindeutige Barcode-Sequenzen an, sogenannte Unique Molecular Identifier (UMI). Theoretisch könnte man die Zellen hierzu dissoziieren und danach mit einem Zellsortierer einzeln in Multiwells platzieren. Anschließend gibt man zu jeder Einzelzelle die Reagenzien für die reverse Transkription zu. Die Poly-A-Schwänze der mRNA adressiert man mithilfe von Poly-T-Primern, die durch einen UMI verlängert sind. Auf diese Weise erhält jede Zelle einen individuellen Barcode für die eigenen mRNAs. Isoliert man anschließend die cDNA, weiß man nach dem Sequenzieren anhand des UMI, zu welcher Zelle jede Sequenz gehört. Besonders effizient ist diese Herangehensweise aber nicht. Selbst ein Organismus wie Caenorhabditis elegans dürfte mit seinen nur rund tausend Zellen jeden Experimentator beim Pipettieren in den Wahnsinn treiben. Zumal er pro Wurm eintausend UMIs bräuchte.
Um dieses Problem zu lösen, haben sich Junyue Cao et al. von der University of Washington vor fünf Jahren einen Trick ausgedacht – und diesen auch an C. elegans sowie einigen Säugerzelllinien erprobt (Science 357(6352): 661-7).
In der ersten Runde werden die Zellen nur grob auf die Wells einer Mikrotiterplatte verteilt. Für die reverse Transkription befinden sich also mehrere Zellen im selben Näpfchen. Bei einer 96-Well-Platte sind dazu 96 Barcodes nötig, falls jedes Well befüllt wird. Danach mischt man die Zellen wieder und verteilt sie mithilfe eines Zellsortierers erneut auf einer Multiwell-Platte. Das Gerät wird so eingestellt, dass es eine vorgegebene Anzahl von Zellen in jedes Gefäß sortiert – zum Beispiel zehn C.-elegans-Zellen pro Well. Weil die Zellen zuvor durchmischt wurden, ist es unwahrscheinlich, dass zwei Zellen mit identischem Barcode im selben Volumen landen. Anschließend hängt man einen zweiten UMI an die cDNA. Hierfür nutzte die Gruppe ein Verfahren, das eigentlich entwickelt wurde, um die Zugänglichkeit von Chromatin auf Ebene des Gesamt-Genoms zu erfassen. Man verwendet dazu eine Transposase mit verstärkter Aktivität. Die Transposase schneidet die DNA an vielen Stellen und fügt an diesen eine kurze Basenfolge ein, die ebenfalls einen eindeutigen Barcode enthält. Weil Chromatin-reiche Regionen für die Transposase schlechter zugänglich sind, taucht der Barcode vor allem in Abschnitten mit geringer Chromatin-Dichte auf und markiert die Teile des Genoms, die für Transkriptionsfaktoren ablesbar sind. Die Methode nennt sich Assay for Transposase-Accessible Chromatin using sequencing oder kurz ATAC-Seq.
Bei dieser Reaktion wird auch vorhandene cDNA mit Barcodes versehen, die man später in den Sequenzen an ihrer Polyadenylierung erkennt. Für die Einzelzell-Transkriptomik haben die US-Amerikaner die neu kombinierten Zellen mit der Transposase inkubiert. Idealerweise besitzt danach jede Sequenz, die eine mRNA repräsentiert, zwei Barcodes. Hätte man also für die reverse Transkription einhundert UMIs zur Auswahl und für den zweiten Schritt nochmals einhundert, so gäbe es bereits 10.000 verschiedene Kombinationen. Die Wahrscheinlichkeit, dass zwei Transkriptome dieselbe Code-Kombination erhalten, ist gering und geht gegen null, wenn man in einer weiteren Runde einen dritten Barcode zufügt. Die Gruppe nennt diesen Ansatz Single-Cell combinatorial Indexing RNA Sequencing oder etwas eingängiger sci-RNA-seq. Der besondere Kniff ist die kombinatorische Indexierung, mit der vergleichsweise wenige Barcodes ausreichen, um unzählige Zellen eindeutig zu markieren.
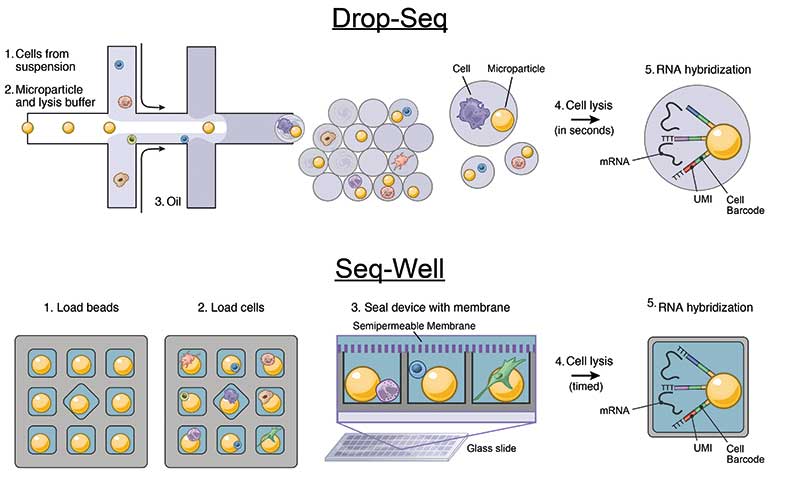
Diese Arbeit muss man sich heute in vielen Fällen aber nicht mehr machen. Inzwischen sind Beads erhältlich, die bereits mit Poly-T-Primern für die reverse Transkription versehen sind – jedem einzelnen Bead ist hierdurch ein einmaliger Barcode zugeordnet. Die Beads kann man auf Nanowell-Chips geben, in deren Vertiefungen nur jeweils ein Bead Platz findet. Danach verteilt man die dissoziierten Zellen über die Chips und startet die reverse Transkription. Die Zellen müssen natürlich so verdünnt sein, dass möglichst nicht mehr als eine Zelle an einem Bead landet. Beim sogenannten Seq-Well-Verfahren ist zum Beispiel ein Array von 86.000 Nanowells auf einem Chip untergebracht (Nat. Methods 14(4): 395–8).
Einfacher mit Tropfengenerator
Beim Pipettieren ist bei diesen Techniken aber ein wenig Fingerspitzengefühl gefragt, um zunächst die Beads und danach die Zellen gleichmäßig auf die Wells zu verteilen. Für weniger geschickte Hände dürften daher Tropfen-basierte Methoden besser geeignet sein. Bei diesen wandern die dissoziierten Zellen oder isolierten Zellkerne durch den Kanal eines Mikrofluidik-Systems. Über weitere Kanälchen münden Beads in den Strom, und aus einer anderen Richtung fließt eine ölige Flüssigkeit hinzu. Durch Phasentrennung entstehen hierdurch kleine Tröpfchen. Wie beim Nanowell-Verfahren muss auch hier sichergestellt sein, dass jedes Tröpfchen maximal ein Bead enthält – und jedem Bead sollte auch nur eine einzige Zelle zugeordnet sein. Zwar kann man eventuell doppelt vergebene Barcodes später bioinformatisch aus den Daten herausrechnen, dennoch muss der Großteil aller sequenzierten Zellen einzeln mit genau einem Bead in einem Tropfen gelandet sein.
Bei vielen Droplet-Systemen erzwingt die Statistik einen verschwenderischen Umgang mit Reagenzien und Material: Die meisten Tropfen sind leer, und Tropfen, die ein Bead enthalten, fehlt meist eine Zelle. Inzwischen bieten Firmen wie zum Beispiel 10x Genomics optimierte Mikrofluidik-Systeme an. Der Zufluss ist so reguliert, dass nahezu jeder Tropfen genau ein Bead abbekommt. Aber auch diese Chips müssen sparsam mit Zellen beladen werden, um „doppelte Belegungen“ einzelner Beads zu vermeiden.
Christoph Bocks Gruppe am CeMM Forschungszentrum für Molekulare Medizin und der Medizinischen Universität Wien entwickelte ein Verfahren, mit dem man durch kombinatorisches Indexing Beads mehrfach beladen und die Sequenzdaten dennoch einzelnen Transkriptomen zuordnen kann. Um dabei auch dem, wie es in der Publikation der Wiener heißt, „eleganten Design [der sci-RNA-Seq] Tribut zu zollen“, nennen sie ihr Verfahren scifi-RNA-Seq, wobei scifi für Single-Cell combinatorial fluidic Indexing steht (Nat. Methods 18(6):635-642).
„Die scifi-RNA-Seq-Methode wurde von Paul Datlinger in meinem Labor entwickelt“, würdigt Bock den Erstautor des Papers. Der habe nach einem Verfahren für mindestens eine Million Zellen gesucht, das zugleich praktikabel und einigermaßen kostengünstig sein sollte. In ihrer Publikation bringt die Gruppe die Ineffizienz herkömmlicher Droplet-Systeme auf den Punkt: „Die meisten Tropfen sind voll funktionsfähig, enthalten Mircrobeads mit Barcodes und die Reagenzien für die reverse Transkription – aber sie stoßen niemals auf eine Zelle und können damit zu keinem Einzelzell-Transkriptom beitragen.“
Überladene Beads
Im Gegensatz zur sci-RNA-Seq kommt die speziell auf barcodierte Beads ausgerichtete scifi-RNA-Seq mit nur einer Pre-Indexing-Runde aus, in der Zellen die reverse Transkription auf einer 96- oder 384-Well-Platte durchlaufen und mit dem ersten UMI markiert werden. Die Zellen werden anschließend wieder vermischt und in die Mikrofluidik eingespeist. Dabei nimmt das Team bewusst ein Überladen der Beads mit mehreren Zellen in Kauf. Den mRNA-Sequenzen wird dann zwar derselbe Barcode über das Bead zugewiesen, sie unterscheiden sich aber sehr wahrscheinlich durch den beim Pre-Indexing vergebenen Identifier.
In diesem Fall findet keine reverse Transkription an den Beads statt, da auch hier die bereits vorhandene cDNA markiert wird. „Deswegen verwenden wir auch nicht den normalen Kit für 10x RNA-Seq, sondern den 10x Kit für ATAC-Seq, den wir quasi zweckentfremden“, erläutert Bock die Reaktion in den Tröpfchen. „Wir haben scifi-RNA-Seq für die 10x-Methode optimiert, allerdings ist sie auch flexibel auf andere Mikrofluidik-Methoden anwendbar wie Droplets und Subnanoliter-Wells.“
Anfangs sei gar nicht klar gewesen, ob die Droplet-Generatoren überhaupt mit derart hohen Zellkonzentrationen klarkommen, erinnert sich Bock. „Wir haben es probiert, und es hat funktioniert.“ Die Ausbeute ließ sich enorm steigern, und die Gruppe kam mit weniger Reagenzien aus. „Wir haben bis zu anderthalb Millionen Zellen, statt der üblichen etwa 10.000 Zellen, auf einen Channel des 10x-Chips geladen, ohne dass es Probleme gab“, so Bock. Er betont aber auch, dass nicht alle Zelltypen für die Technik geeignet sind, etwa wenn sie zu groß oder zu empfindlich sind. Bock rät zudem zu anderen Methoden, wenn man nur wenige Zellen sehr tiefgehend und umfassend analysieren will. Aber: „scifi-RNA-Seq eignet sich besonders für Anwendungen, die Hunderttausende oder Millionen Zellen gleichzeitig analysieren.“ Die Technik ist also immer dann interessant, wenn ein hoher Durchsatz im Vordergrund steht.
Begeistert von der Einzelzell-Analytik ist auch Nikolaus Rajewsky, der am Berliner Institut für Medizinische Systembiologie des Max-Delbrück-Centrums für Molekulare Medizin eine Gruppe leitet. „Die Einzelzell-Methoden ermöglichen eine unvorstellbare Schärfe und Genauigkeit der Daten“, schwärmt er. „Für mich ist das wie ein neues Mikroskop, das es erlaubt, in die Zellen hineinzuschauen, während sie das Buch des Lebens auslesen“. Rajewsky sieht sein Labor zwar zum Teil auf der Seite der Methoden-Entwickler, jedoch immer zielgerichtet, um neuen Fragen in der RNA-Forschung auf den Grund zu gehen. „Wir wenden die Methoden in der Grundlagenforschung an. In den letzten Jahren interessiere ich mich aber sehr für die direkte Anwendung bei Krankheiten.“
Zelle ist nicht gleich Zelle
Rajewsky betont, dass es tatsächlich einen Unterschied macht, wenn man das Transkriptom einzelner Zellen in den Daten unterscheiden kann und nicht das Gewebe oder das Organ einfach als Ganzes sequenziert. „Die Zellen der Niere sind eben nicht alle gleich“, nennt er ein Beispiel. „Da gibt es riesige Unterschiede und verschiedene Zelltypen. Die müssen sich absprechen und organisieren, und diese Funktionen können wir jetzt molekular verstehen“.
Daten aus Einzeltranskriptom-Analysen kann man in Plots visualisieren, die einzelne Zellen als Punkte darstellen. Die Punkte liegen aber nicht in einem dreidimensionalen Raum: Sie haben unter Umständen viele Tausend Koordinaten, die jeweils für abgelesene Gene stehen, die mithilfe der mRNA erfasst werden. Mit Verfahren zur Dimensions-Reduktion lassen sich die Ergebnisse dennoch zweidimensional veranschaulichen. Zellen mit ähnlichen Expressions-Profilen sind dann als Wolken und Cluster zu erkennen. Die Auswertung und Interpretation der Daten ist aber nicht trivial. In dem Sequenz-Datensatz einer gesamten Zellpopulation (Bulk-Sequencing) mitteln sich Ausreißer heraus. Bei einer einzelnen Zelle weiß man zunächst jedoch nicht, ob sie sich bei einer auffälligen Abweichung wirklich von den anderen unterscheidet oder nicht. „Man kann das Experiment ja schlecht wiederholen, weil es diese Zelle ja nicht mehr gibt“, beschreibt Rajewsky das Dilemma.
Andererseits soll die Einzelzell-Transkriptomik gerade auch seltene Zellpopulationen sichtbar machen. „Man bekommt noch immer nicht das komplette Transkriptom pro Zelle“, räumt Rajewsky ein. „Wahr ist, dass man schwach exprimierte Gene häufig nicht sieht, und das ist nach wie vor ein Problem.“ Dennoch nähere man sich immer mehr der Realität an, weil es auch alternative Methoden gibt, mit denen sich Einzelzell-Transkriptome vergleichen lassen. „Im Kommen sind Single- Molecule-Imaging-Methoden, bei denen man Zehntausende Sonden auf Target-mRNAs in Zellen und Geweben hybridisieren kann; das ist dann eine komplementäre Methode zum Sequencing-Approach.“
Vergleich mit Bekanntem
Oder man orientiert sich an bereits bekannten Prozessen und verifiziert mit diesen die Ergebnisse der Transkriptom-Analyse. Rajewsky erwähnt eine Arbeit seiner Gruppe in Science (358(6360): 194-9). „Da haben wir einen Fliegenembryo auf Einzelzell-Ebene räumlich rekonstruiert.“ Das Team hatte Drosophila-Embryonen in einem 6.000-Zellstadium untersucht und die Expression von rund 8.000 Genen auf Einzelzell-Ebene erfasst. Mit einem speziell für die räumliche Rekonstruktion entwickelten Algorithmus entstand daraus ein dreidimensionales Modell des Embryos. „Dazu haben wir dann auch In-situ-Hybridisierungen durchgeführt, um uns davon zu überzeugen, dass das stimmt, was wir gemessen haben.“
Auch das Vorbereiten der Zellen für die Analytik kann sich auf das mRNA-Profil auswirken und Ergebnisse verfälschen. „Beim Dissoziieren stressen Sie die Zellen. Damit sind die Single-Cell-Daten kontaminiert. Deswegen sequenziert man häufig lieber einzelne Zellkerne. Dafür muss man das Gewebe nicht mehr dissoziieren, sondern kann die Nuklei aus einer Art Brei heraus isolieren.“
Weil die räumliche Information bei den meisten Einzelzell-RNA-Sequenziermethoden verloren geht, muss man, wie oben erwähnt, auf Software-gestützte Techniken zurückgreifen, um rückblickend wahrscheinliche Nachbarschaften zwischen den Zellen zu ermitteln. Für Untersuchungen an Organoiden verwendet Rajewskys Gruppe Methoden, die auch die räumlichen Informationen der Transkripte erfassen. „Wir können einen Gewebeschnitt auf eine Oberfläche packen, die mit einigen biochemischen Tricks so beschaffen ist, dass die RNA aufgefangen wird und einen Barcode aus Oligonukleotiden bekommt“, erklärt Rajewsky. Die Barcodes enthalten dann eine Information über die Position auf der Fläche. „Mit Kryostaten kann man sehr feine Gewebeschnitte anfertigen, wir arbeiten im Moment mit einer Dicke von etwa zehn Mikrometern – das entspricht ungefähr einer Einzelzellschicht.“
Räumliches Transkriptom
Das Verfahren will Rajewsky Computergestützt verfeinern, um Einzelzell-Transkriptome aus mehreren Gewebeschnitten zu einem dreidimensionalen Bild rekonstruieren zu können (mehr zur räumlichen Transkriptomik finden Sie im Methoden-Special in LJ 4/2021 ab Seite 60, Link).
Rajewsky ist aber nicht nur an mRNA interessiert, sondern auch an nicht-codierenden RNAs. „microRNAs werden fast nie in einzelnen Zellen sequenziert“, stellt er fest, „es sind ein paar Protokolle dazu publiziert worden, aber ich glaube, da gibt es noch viel zu tun.“
2021 stellten Alina Isakova, Norma Neff und Stephen Quake von der Stanford University die Smart-Seq-total-Technik vor, mit der es möglich ist, neben der mRNA auch lange nicht-codierende RNAs und microRNAs in einzelnen Zellen zu sequenzieren (PNAS 118(51): e2113568118). Die Gruppe verwendet dazu die MMLV-Reverse-Transkriptase, die einzelsträngige Nukleinsäuren um einen komplementären DNA-Strang ergänzt – unabhängig davon, ob sie einen Poly-A-Schwanz tragen oder nicht.
Auch Rajewskys Team arbeitet an der Sequenzierung von microRNAs und nicht-codierenden RNAs in einzelnen Zellen. Rajewsky kann aber derzeit noch nicht über Details sprechen. „Wir glauben, dass wir da Fortschritte gemacht haben, und was ich schon verraten darf: Wir können microRNAs und mRNAs aus derselben Zelle sequenzieren.“ Für Rajewsky würde das eine neue Tür öffnen: „Dann könnten wir sehen, was eine microRNA in der einzelnen Zelle macht, denn microRNAs binden ja an mRNAs und regulieren deren Expression. Das wird noch mal sehr spannend!“