Herkunftsnachweis mit Zellstammbaum - Cell Lineage Tracing
Mario Rembold
(10.12.2021) CRISPR-Cas9 und Einzelzell-Sequenzierung helfen Entwicklungsbiologen und Krebsforschern, Zellstammbäume zu rekonstruieren. Die Sache funktioniert aber nur, wenn die Algorithmen der Analysesoftware die Eigenheiten der Einzelzell-Sequenzierung berücksichtigen und während der Aufarbeitung verlorene RNA einkalkulieren.
Vor fast 40 Jahren saßen der spätere Nobelpreisträger John Sulston und seine Kollegen vom Medical Research Council Laboratory of Molecular Biology in Cambridge, UK, über Stunden geduldig vor dem Mikroskop. Sie sahen einer Zygote dabei zu, wie sie sich das erste Mal teilte, wie daraus ein Embryo und schließlich ein kompletter Wurm entstand. Mit bloßem Auge am Okular dokumentierte das Team, wie es mit jeder einzelnen Zelle weiterging. Das Kurzzeitgedächtnis des Beobachters sei dabei ein limitierender Faktor, resümierten sie später in ihrem Artikel, der 1983 in Developmental Biology erschien (100(1): 64-119).
Erstmals hatten die Wissenschaftler darin die Entwicklung von Caenorhabditis elegans akkurat dokumentiert und ein „hochgradig invariantes“ Schicksal der einzelnen Zelllinien erkannt.
C. elegans gehört bis heute zu den Lieblingsorganismen der Entwicklungsbiologen, nicht zuletzt weil er sich so gut beobachten lässt. Dennoch dürfte kaum jemand die Forscher von damals beneiden.
Tatsächlich war das Auge in Kombination mit der Lichtmikroskopie über Jahrzehnte das beste Instrument, um in einem Embryo das Schicksal der Tochterzellen zu verfolgen. Farbstoffe halfen zwar, die richtigen Zellen nicht aus den Augen zu verlieren, doch mit jeder Teilung verringerte sich auch die Konzentration des Farbstoffs.
Alternativ verwendete man später Zellen aus genetisch modifizierten Individuen, die zum Beispiel GFP synthetisierten, und deren Zellen immer gleich hell leuchteten. Die Forscher schleusten eine oder mehrere embryonale Stammzellen eines solchen Tieres in den unmarkierten Embryo ein und erhielten einen chimären Organismus. Allerdings konnten sie nie ausschließen, dass sich der Embryo durch das Transplantat anders verhielt, oder die eingebrachten Zellen ihrerseits geringfügig andere Prozesse durchliefen.
Heute ist es dank Gene Editing viel leichter, das Schicksal von Zellen anhand eindeutiger genomischer Barcodes zu verfolgen: Anstatt tagelang durchs Mikroskop zu starren, pickt sich der Experimentator den Embryo oder den fertig entwickelten Organismus einfach in einem bestimmten Stadium heraus und ermittelt die Herkunft jeder einzelnen Zelle rückblickend mithilfe eines Zellstammbaums. So kann er das Schicksal einer Zelllinie im Idealfall von der einzelligen Zygote bis zum fertig entwickelten Organismus verfolgen – man spricht daher auch von Cell Lineage Tracing.
Bastiaan Spanjaard, eigentlich Theoretischer Physiker, erstellt solche Zellstammbäume und rekonstruiert die embryonale Geschichte einzelner Zellen. Zunächst war Spanjaard einige Jahre in Alexander van Oudenaardens Team an der Universität Utrecht, inzwischen arbeitet er in Berlin am Max-Delbrück-Centrum für Molekulare Medizin im Labor von Jan Philipp Junker – der zuvor ebenfalls in der AG van Oudenaarden geforscht hatte.
Türöffner Einzelzell-RNA-seq
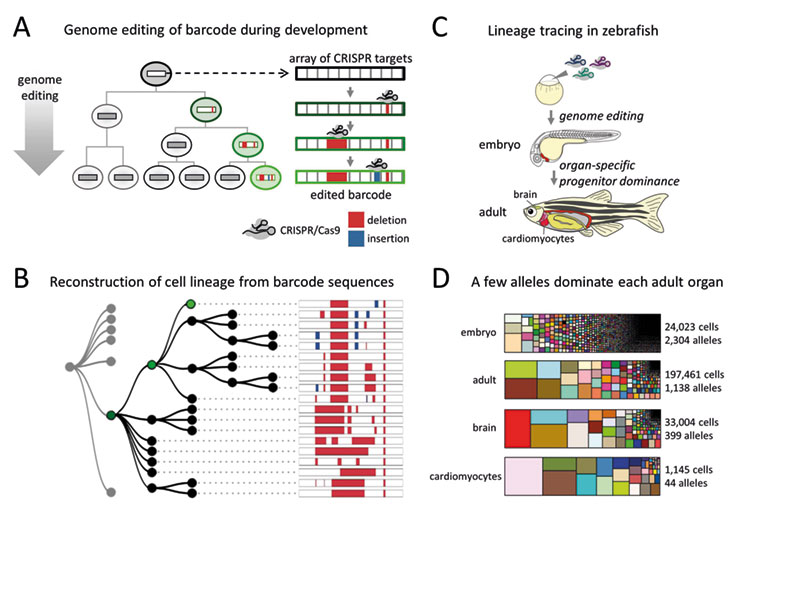
Die niederländische Gruppe und die aus ihr hervorgegangenen Postdocs trugen in den vergangenen Jahren zu diversen neuen Techniken des Cell Lineage Tracings bei. „Die Einzelzell-Sequenzierung hat die Entwicklung sehr vorangebracht“, erwähnt Spanjaard den vielleicht wertvollsten Türöffner für neue Herangehensweisen und nennt als weitere wichtige Köpfe der Szene den Direktor des Basler Biozentrums Alex Schier, der gleichzeitig an der Harvard University eine Gruppe leitet, sowie Jay Shendure von der Universität Washington. Die Teams um Shendure und Schier hatten 2016 eine CRISPR-Cas9-basierte Methode vorgestellt, mit der man einzelne Zelllinien mit individuellen Barcodes versehen kann (Science 353(6298): aaf7907; siehe auch laborjournal.de/editorials/1061.php).
Das Verfahren nannten die Autoren GESTALT – für Genome Editing of Synthetic Target Arrays for Lineage Tracing. Fügt man in einem bestimmten Stadium Mutationen in das Genom einzelner Zellen ein, so tragen auch alle Nachkommen dieser Zellen die Mutation. Sequenziert man die einzelnen Zellen, findet man heraus, in wie vielen Zellen diese Mutation vorhanden ist – und auf welchen gemeinsamen Vorläufer diese Zellen zurückzuführen sind.
Allerdings waren Cell-Lineage-Tracing-Methoden vor CRISPR-Cas9 vom Zufall geprägt. Wo die Mutation lokalisiert war, wusste man nicht. Die Genschere Cas9 wird hingegen mithilfe von guide-RNAs zu ausgesuchten Loci im Genom geführt und induziert dort durch Schnitte auf der DNA an einem genau bekannten Ort zufällige Mutationen. Der Forscher weiß also beim Sequenzieren, wo die Markierung liegen sollte – er muss nicht mehr auf gut Glück das gesamte Genom aller Zellen sequenzieren, um die Mutation zu finden.
Bei GESTALT wird ein Barcode erzeugt, indem die Genschere auf einem bekannten kurzen Abschnitt zufällig mehrere Basen abändern kann, aber nicht muss. Dieses Array nahe beieinander liegender möglicher CRISPR-Targets umfasst rund zehn verschiedene Basen, an denen es im Lauf der Embryonalentwicklung zu Änderungen kommen kann. Statistisch sind damit hunderttausende unterschiedliche Barcodes möglich.
Vererbter Barcode
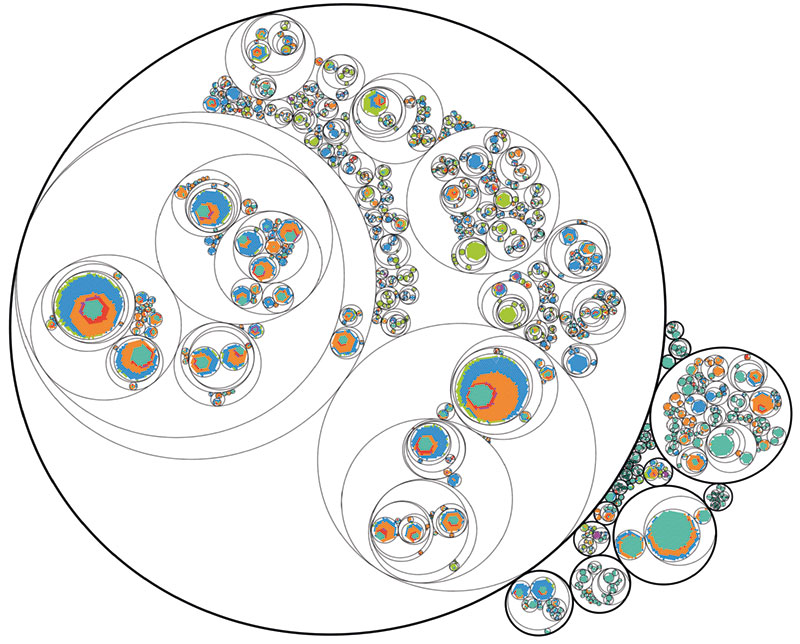
„Wenn man im Einzell-Stadium solch einen Barcode generiert, findet man ihn später in allen Zellen wieder“, erklärt Spanjaard. Auch andere Methoden zum Cell Lineage Tracing setzen heute vor allem auf CRISPR-Cas9 und nutzen vergleichbare Markierungsstrategien. Man kann die Barcodes so designen, dass eine einmal geschriebene „Ziffer“ kein zweites Mal von der Genschere gefunden und geschnitten werden kann. In Nachkommen dieser Zelle können aber an anderen Positionen des Arrays neue Mutationen und damit neue Barcodes generiert werden. Mit der Zeit akkumulieren Mutationen am Barcode, und man erhält im entstehenden Embryo verschiedene unterschiedlich getaggte Zellen. Ein Stück weit ist die Idee vergleichbar mit einer fortlaufenden Nummer, die die Nachkommen einer Zelllinie weiterführen. „Wenn eine bestimmte Markierung in der Hälfte der Zellen zu sehen ist, dann gehen diese alle auf das Zweizell-Stadium zurück“, nennt Spanjaard ein Beispiel. Über die Barcode-Analyse lässt sich also ein Stammbaum aller von der Genschere markierten Zellen erstellen.
„In den letzten drei Jahren wurden Lineage Tracing und Einzelzell-Sequenzierung mit der Transkriptomik verbunden“, nennt Spanjaard einen weiteren Meilenstein. Hier kommen auch neue Analysetools für den Computer ins Spiel, die an diese Herausforderungen angepasst sind. „Das ist mein Tätigkeitsfeld: Ich schreibe Software“, geht Spanjaard auf seinen Arbeitsalltag ein. „Als Entwickler darf man die Algorithmen aber nie als Blackbox sehen“, mahnt er, „man muss sich vergewissern, dass die Analysen auch Sinn ergeben“.
Wichtige Validierung
Ein Programm validieren könne man unter anderem mithilfe simulierter Daten, die echten Transkriptomik-Datensätzen ähneln, bei denen der virtuelle Stammbaum jedoch bekannt ist. „Dann testet man, ob der eigene Algorithmus auch wirklich diesen Stammbaum herausbekommt.“ Weiterhin könne man in echten Datensätzen schauen, ob der daraus ermittelte Baum auch kompatibel ist mit Ektoderm, Mesoderm und Endoderm, denen man jede Zelllinie letztlich zuordnen kann.
„Eine andere Verifizierung, die ich sehr schön finde, ist der Blick auf die Immunzellen“, fährt Spanjaard fort. Diese kann man via Zellsortierung aus verschiedenen Organen isolieren und dann einzeln sequenzieren. „Dann sieht man, dass die Immunzellen im Stammbaum immer zusammen auftauchen“, erklärt er. „Ich kann das Gehirn und das Herz eines Individuums sequenzieren und stelle fest: Das Gehirn geht auf das Ektoderm zurück, das Herz auf das Mesoderm – aber die Immunzellen aus Gehirn und Herz haben eine gemeinsame Herkunft.“ Ein brauchbares Software-Tool sollte also die Immunzellen, die auf unterschiedliche Organe verteilt sind, auf demselben Abzweig des Zellstammbaums platzieren.
Großer Teil RNA geht verloren
Deep Learning mithilfe neuronaler Netze setzt Spanjaard derzeit in seinen Algorithmen noch nicht ein. „Irgendwann wird so etwas aber sicher kommen“, blickt er in die Zukunft. Umgekehrt kann er aber auch nicht einfach auf die klassischen Analysetools zurückgreifen. „Diese Methoden sind für Einzelzell-Dateien nicht gut geeignet“, begründet er, „denn man hat mit ihnen ein Ausfallproblem.“ Die Software der alten Schule baut auf Datensätzen auf, die aus einer Sammlung vieler Zellen hervorgehen. In solchen Bulk-Analysen wird ein Großteil der mRNA zu cDNA transkribiert und dann sequenziert. In einer einzelnen Zelle geht aber ein erheblicher Teil der RNA durchs Netz. Manchmal sind lediglich zehn Prozent der mRNA-Sequenzen in den Datensätzen einer Einzelzelle enthalten. „Außerdem erfasst man nur rund ein Viertel aller Barcodes in den Zellen“, ergänzt Spanjaard. Auf all das muss ein Analysetool optimiert sein, wenn es für modernes Cell Lineage Tracing verwendet wird.
Nur geringe zeitliche Auflösung
Über genomische Barcodes kann man die Herkunft einer Zelle rekonstruieren und mit dem Transkriptom abgleichen. Dabei lernt man auch etwas über Änderungen der Genexpression während der Musterbildung und Differenzierung. Allerdings ist die zeitliche Auflösung sehr grob und indirekt. Zwei Zellpopulationen können zum Beispiel aus einem Vorläufer hervorgehen. Dann gibt der Vergleich der Transkriptome Aufschluss darüber, was bei der Differenzierung anders gelaufen ist. Doch welches Gen zum aktuellen Zeitpunkt herauf- oder herunterreguliert wird, sieht man nicht. Vielleicht liegt eine mRNA in hoher Konzentration vor, doch eigentlich ist das wesentliche Merkmal dieser Zellen, dass die mRNA gerade abgebaut wird.
Um zeitlich hochaufgelöste Informationen des Transkriptoms zu erhalten, benötigt man einen anderen Trick. Gioele La Manno et al. stellten 2018 hierfür das Konzept der RNA-Velocity vor (Nature 560(7719): 494-8). Aus dem Verhältnis von reifer zu ungesplicter mRNA eines Transkripts lässt sich abschätzen, ob die mRNA netto gerade entsteht oder abgebaut wird. Natürlich hat man auch in diesem Fall nur einen statischen Schnappschuss des Transkriptoms, aber mit einer zeitlichen Ableitung. So kann man einordnen, ob zuvor mehr oder weniger reife mRNA zur Verfügung stand. Man bekommt also einen Eindruck davon, was die Zelle in diesem Moment an ihren Transkripten verändert.
Interessiert sich ein Entwicklungsbiologe hingegen für die räumliche Verteilung der Geschehnisse im Embryo, geht diese Information komplett verloren, wenn man die Zellen vereinzelt und ihre Transkriptome ermittelt. Und klassische Methoden wie die In-situ-Hybridisierung mit Sonden gegen ausgewählte mRNA belassen alle anderen Transkripte im Dunkeln. Man muss also vorher genau wissen, was man anschauen will – ein unvoreingenommener Blick auf das gesamte Transkriptom ist nicht möglich.
Um die räumliche Verteilung der Transkripte zu bewahren, entwickelte Junker während seiner Zeit in Utrecht die sogenannte Tomo-Seq-Technik. 2014 stellte die Gruppe die Methode vor, 2016 veröffentlichte sie ein ausführliches Protokoll in Methods in Cell Biology (135: 299-307). Das Verfahren wurde für Zebrafische etabliert: Embryonen oder auch einzelne Organe werden tiefgefroren in dünne Scheibchen geschnitten und später für die Einzelzell-Sequenzierung präpariert. „Heute ist die Methode so fortgeschritten, dass man in der Scheibe zusätzlich x- und y-Koordinaten für die einzelnen Spots aufzeichnen kann“, berichtet Spanjaard. Man erhält also eine Karte mit der dreidimensionalen Verteilung der Transkripte.
Hat man eine grobe Idee von zeitlicher Dynamik und räumlicher Verteilung der mRNAs, kann man gezielt ein interessant erscheinendes Zielmolekül auswählen, etwa für klassische Methoden wie In-situ-Hybridisierung, Antikörperfärbung oder einen fluoreszierenden Reporter.
Über Umwege ohne Barcode
Zelllinien zu verfolgen ist nicht nur hilfreich, um die Embryonalentwicklung zu verstehen. Auch im voll entwickelten Organismus teilen sich noch Zellen – das ist vor allem dann relevant, wenn etwas nicht ordnungsgemäß funktioniert, etwa bei der Krebsentstehung. Im menschlichen Organismus kann man aber nicht auf Barcodes zurückgreifen. Hier muss man altmodischer vorgehen und nutzt den Umstand, dass natürlich auftretende somatische Mutationen auch an die Tochterzellen weitervererbt werden. Der Sequenzier-Aufwand ist dadurch zwar höher, die Möglichkeiten der Einzelzell-Transkriptomik kann man aber genauso anwenden, etwa um Biopsien von Patienten zu untersuchen.
2018 wirkte Spanjaard als Erstautor an einem Verfahren zur Rekonstruktion von Zellstammbäumen mit. Die Technik nennt sich LINNAEUS, was für Lineage Tracing by Nuclease-Activated Editing of Ubiquitous Sequences steht (Nat. Biotechnol. 36(5): 469-73). Auch hier geht es um die gleichzeitige Einzelzell-Transkpriptomik tausender Zellen – das Team hatte per CRISPR-Cas9 Markierungen im Genom hinterlassen, um Zellen später einer Linie in einem Stammbaum zuordnen zu können. Als Modell nutzten die Forscher den Zebrafisch, und schauten sich auch Organe erwachsener Tiere an, unter anderem das Herz.
Rätselhafter Zelltyp
„Auch im späteren Leben des Fisches liefern die Zellstammbäume noch ziemlich interessante Informationen“, ordnet Spanjaard die Ergebnisse ein. Aktuell untersucht er mit ihnen die Herzregeneration beim Zebrafisch. „Dabei sehen wir einen Zelltyp, der im Herz eines gesunden unverletzten Fisches nicht existiert“, so Spanjaard. Diese Zellen stammen offensichtlich nicht aus einem Stammzellreservoir: „Wahrscheinlich findet eine Rückdifferenzierung statt, denn eine Stammzellpopulation haben wir dort nie gefunden.“ Wenn die Wundheilung abgeschlossen ist, verschwinden diese Zellen wieder – das sei nach rund dreißig Tagen der Fall.
„Schauen wir uns die Herkunft dieser Zellen an, sehen wir, dass einige von der äußeren Seite des Herzens einwandern, andere von der Innenseite.“ Die Zellen aus dem Herzinneren seien vor allem bei tieferen Wunden zu sehen. „Das Beeindruckende ist: Die beiden Zelltypen sehen transkriptomisch sehr ähnlich aus“, berichtet Spanjaard. „Ohne die Lineage-Tracing-Information würde man sagen: Die Expression zeigt so hohe Übereinstimmungen, dass die Zellen denselben Ursprung haben müssen.“ Vorläufige Ergebnisse hierzu stellen Spanjaard und seine Kollegen aktuell auf bioRxiv zur Diskussion (doi: 10.1101/2021.01.07.425670).
Das Beispiel zeigt, dass nicht alles, was gleich aussieht, auch gleich ist – oder unterschiedliche Zellen auf eine gleiche Funktion umschalten können.
Die Genschere CRISPR-Cas ist ein effektives Werkzeug für die Herstellung individueller Cell-Lineage-Tracing-Barcodes. Von solch einer Hilfe konnten frühere Forscher nur träumen – als sie am Mikroskop Lidschlag um Lidschlag unterdrückten, um das weitere Schicksal der untersuchten Zelle bloß nicht aus den Augen zu verlieren.