Mutationen auf der Spur
SARS-CoV-2-Methoden: Varianten-Detektion ohne NGS
Andrea Pitzschke
(09.06.2021) In der letzten LJ-Ausgabe beschrieb Sascha Tierling vom Institut für Genetik/Epigenetik der Universität des Saarlandes eine von ihm entworfene Primer-Extension-Technik zur schnellen Detektion von SARS-CoV-2-Varianten. Findige Forscher entwickelten für die Varianten-Analyse aber noch weitere, teils sehr unterschiedliche Methoden.
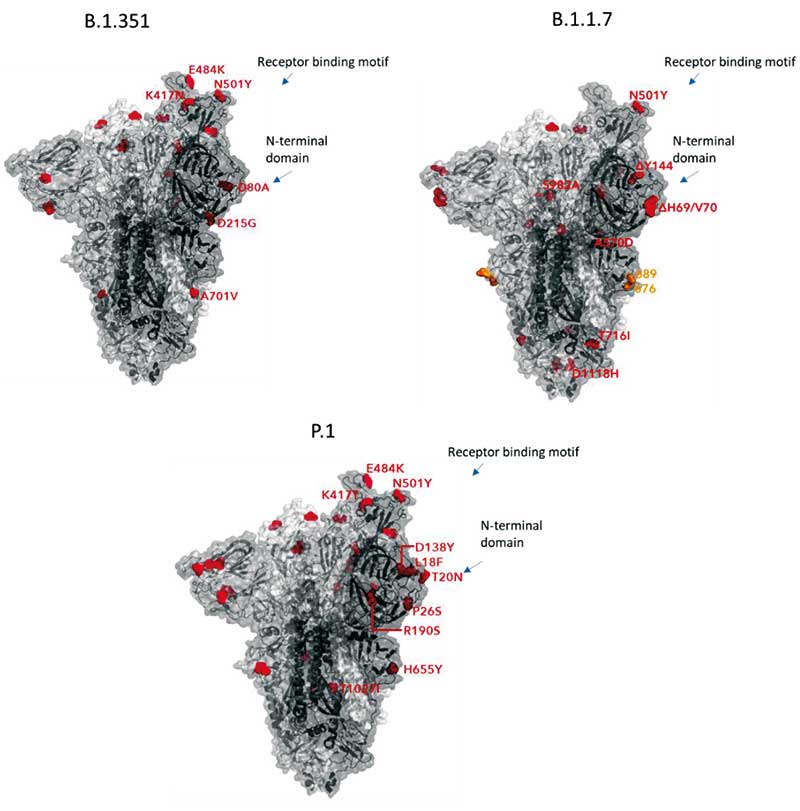
Mutationen bei SARS-CoV-2 sind zwar seltener als bei Influenzaviren, aber dennoch zum Teil besorgniserregend. Sogenannte Variants of Concern (VOC) übertragen sich effizienter, haben eine höhere Wahrscheinlichkeit für Re-Infektionen, ändern den Krankheitsverlauf oder das Altersspektrum, dämpfen die Wirksamkeit von Impfstoffen oder verursachen falsch-negative Testergebnisse. Spätestens seit Auftreten der Abstammungslinien B.1.1.7, B.1.351, P.1 und zuletzt B.1.617.2 (indische Variante) ist klar, dass Tests nicht nur den bloßen Infektionsstatus feststellen sollten, sondern zumindest in Stichproben auch die vorliegende Virus-Variante.
Die komplette Sequenzinformation eines Virus erhält man nur, wenn man das vollständige Genom sequenziert (Whole Genome Sequencing). Aufgrund der hohen Kosten kann aber nur ein Bruchteil der positiven SARS-CoV-2-Proben gleichzeitig auch sequenziert werden. Zudem geben die meist erst nach Tagen vorliegenden Sequenzier-Ergebnisse den Virus-Varianten einen Vorsprung, der nur schwer wieder aufzuholen ist. Die derzeit kursierenden VOCs tragen Mutationen primär im Spike-Protein. Man kann sich deshalb auf das S-Gen oder bestimmte Abschnitte daraus beschränken und deren PCR-Produkte mit der Sanger-Sequenzierung analysieren. Die WHO empfiehlt, zur Unterscheidung gängiger Varianten zumindest den N-Terminus codierenden Teil des S-Gens zu sequenzieren, inklusive der Rezeptorbindungsdomäne (aa 1-541) – oder besser noch etwas darüber hinaus (aa 1-800).
Kein PCR-Produkt
Die britische Variante B1.1.7 ist leicht an sechs fehlenden Nukleotiden (nt 207-212) zu erkennen. Sie führen dazu, dass Reverse-Transkriptase-qPCRs (RT-qPCR) fehlschlagen, die auf einen Sequenzabschnitt des S-Gens zielen. Versucht man mit der RT-qPCR im Multiplex-Prinzip Abschnitte von S-Gen, ORF1 und N-Gen zu amplifizieren, liefern nur die letzten beiden PCR-Produkte. Durch die fehlende Amplifikation des S-Gen-Abschnitts (S-Gene Target Failure) outet sich die Deletionsvariante. Ist die Prävalenz von B.1.1.7 hoch, ist die Zuordnung relativ klar. Ob aber womöglich doch eine andere mutierte Variante vorliegt, verrät nur die vollständige Sequenzanalyse.
Schnelle Informationen zu Virus-Varianten erhält man auch mit Schmelzkurven-Analysen der PCR-Produkte. Je nach Mutation zeigt die Schmelzkurve eine charakteristische Verschiebung des Schmelzpunktes. So kam zum Beispiel eine Ende Januar vom Robert-Koch-Institut durchgeführte Ad-hoc-Erhebung zur Verbreitung von B.1.1.7 mithilfe von Schmelzkurvenanalysen zu dem Ergebnis, dass 6,9 Prozent von 1.800 Positiv-Proben die Mutation N501Y sowie die Deletion delH69/V70 aufwiesen, die typisch sind für B.1.1.7.
Einzelnukleotid-Polymorphismen (SNPs) sind ebenfalls ein guter Ansatzpunkt, um bekannte Varianten nachzuweisen. Kennt man die Position und Art des SNPs, lässt sich eine positive Probe ganz gezielt analysieren. Sobald das Virus jedoch eine neue Mutation aufweist, muss auch die SNP-Detektion nachjustiert werden. Für die SNP-Genotypisierung definierten britische Forscher anhand umfassender Sequenzvergleiche spezifische Marker (PLoS ONE 16(2): e0243185).
Die Gruppe konzentrierte sich in den bis zum September 2020 vom COVID-19-Genomics-UK-(COG-UK)-Konsortium zusammengetragenen etwa 40.000 Sequenzen auf polymorphe Allele, die mindestens mit einer Häufigkeit von einem Promille auftraten. Seltenere Polymorphismen wertete sie als identisch nach dem Motto, „die Mutation hat sich nicht durchgesetzt und ist vernachlässigbar“. 41 SNPs erfüllten dieses Kriterium, von denen 22 für eine klare Zuordnung zu einer bestimmten Variante ausreichten.
Daraufhin designten die Forscher Varianten-spezifische Vorwärts-Primer für die Genotypisierungs-Reaktion, die sie mit der sogenannten One-Step-PACE-RT (PCR Allele Competitive Extension) durchführten. Die Vorwärts-Primer tragen am 5´-Ende eine spezielle Tail-Sequenz, die zu einer von zwei FRET-Kassetten im Mastermix passen: Eine ist mit dem gelöschten (gequenchten) Fluoreszenz-Farbstoff FAM gelabelt, die andere mit dem gequenchten Fluoreszenz-Farbstoff HEX. Auf die Tail-Sequenz folgt ein zur Virussequenz passender Abschnitt von fünfzig Nukleotiden, der unmittelbar vor dem SNP liegt. Das 3‘-Nukleotid entspricht einem bestimmten SNP beziehungsweise der Wildtypsequenz. Der universelle Rückwärts-Primer zielt dagegen auf eine konservierte Region fünfzig Nukleotide stromabwärts des SNPs.
Nach der reversen Transkription der Virus-RNA binden die Vorwärts-Primer an die einzelsträngige DNA und werden verlängert. Das geschieht aber nur, wenn das 3‘-Nukleotid passt. Im Verlauf der PCR werden die Tail-Sequenzen in die amplifizierten Fragmente integriert und schließlich auch die zu diesen komplementären Sequenzen hergestellt. Die FRET-Kassette bindet an die passende komplementäre Tail-Sequenz, wodurch die Löschung des Farbstoffs aufgehoben wird und die Probe rot beziehungsweise blau fluoresziert. Sind in der Probe beide Varianten (SNP und Wildtyp) vorhanden, entsteht ein gemischtes Farbsignal. Das Verfahren ist mit entsprechenden 1.536-Well-Platten-Thermocyclern sowie Mikroplatten-Lesegeräten für den Hochdurchsatz geeignet, die Kosten pro Probe liegen unter zwei Euro. Nicht alle Sequenzregionen, die von der Position der jeweiligen SNPs vorgegeben sind, eignen sich jedoch als Marker. In diesen Fällen bleibt das Signal aus.
Gefakte Oligos
Weniger kompliziert klingt dagegen ein ebenfalls auf speziellen Oligonukleotiden beruhendes Verfahren, das kalifornische Forscher von der Krebsdiagnostik abgeschaut haben. Die Molecular-Clamping-RT-qPCR nutzt „gefakte“ Oligos, sogenannte Xeno-Nukleinsäuren (XNAs), die sich über Festphasenpeptidsynthese aus Monomeren von Nukleotid-Imitaten (Fmoc-A,-T,-C,-G) herstellen lassen. Wie klassische dsDNA bilden XNAs Doppelhelices mit Watson-Crick-Basenpaarung, sie paaren sich aber auch mit DNA zu XNA/DNA-Heteroduplexen. XNA/DNA-Doppelstränge sind um einiges stabiler als entsprechende dsDNAs – solange keine Fehlpaarungen existieren. Die aufgrund von Mismatches entstehenden Unterschiede der Schmelztemperatur macht sich die Molecular-Clamping-Methode zunutze: Im Gegensatz zu Mutanten binden Wildtyp-Templates so stark an die dazu passenden XNAs, dass die Amplifikation verhindert wird.
Die Forscher konzentrierten sich auf die Mutationen N501Y sowie D614G und legten zunächst die Größe der Amplifikationsprodukte fest. Dazu wählten sie die Primer für möglichst hoch konservierte Abschnitte so, dass relativ kurze PCR-Produkte (70-150 bp) entstehen. Die XNAs für die Varianten N501Y beziehungsweise D614G decken den Sequenzabschnitt mit der Mutation ab, passen aber exakt zur Sequenz des Wildtyps. Bei der Analyse von Patientenabstrichen mit der One-Step-RT-qPCR setzt man einem Teil der Reaktionsansätze eine XNA zu. RT-qPCR-Ansätze ohne XNA liefern identische qPCR-Kurven, unabhängig von der in der Probe vorhandenen Virus-Variante. Mit XNA entstehen in Proben, die nur den Wildtyp enthalten, praktisch keine detektierbaren PCR-Produkte – im Gegensatz zu Proben mit N501Y oder D614G (medRxiv, doi: 10.1101/2021.04.01.21254484).
Variantensuche mit CRISPR ...
Eine elegante auf CRISPR-Cas basierende Technik zur Detektion von SARS-CoV-2-Varianten entwickelte die Gruppe des indischen Molekularbiologen und Sitar-Virtuosen Debojyoti Chakraborty am CSIR-Institute of Genomics & Integrative Biology in New Delhi. Chakraborty, der am Max-Planck-Institut für molekulare Zellbiologie und Genetik in Dresden bei Frank Buchholz promovierte, nutzt dazu eine spezielle Eigenschaft der Nuklease FnCas9 aus dem Bakterium Francisella novicida: FnCas9 bindet nicht an Zielsequenzen, wenn die eingesetzten sgRNAs an den Positionen 2 und 6, die auf das Proximal Adjacent Motif (PAM) folgen, Mismatches tragen.
Auch andere Mismatches führen zu einer schwächeren Bindung oder verhindern sie ganz. Dies kann man für das Design von sgRNAs ausnutzen, die SARS-CoV-2-Varianten aufspüren. Die Inder durchforsteten die 32 wesentlichen Einzelnukleotid-Varianten von B.1.1.7, B.1.351 sowie P.1 und fanden zehn, die sich als Ziele für entsprechend designte sgRNAs eignen – darunter die in allen drei Varianten vorkommende Mutation N501Y.
Die sgRNAs setzten sie schließlich in einem Rapid Variant Assay (RAY) ein (medRxiv, doi: 0.1101/2021.02.01.21250900). Bei diesem hängt man mithilfe eines biotinylierten RT-PCR-Primers eine Biotin-Gruppe an die amplifizierten Sequenzen von Wildtyp oder Mutanten. Anschließend inkubiert man die PCR-Produkte mit FnCas9 sowie einer auf die gesuchte Mutation zugeschnittenen sgRNA und analysiert die entstehenden Fragmente mit der Agarose-Gelelektrophorese.
Noch flotter und auch außerhalb des Labors kann man die biotinylierten Amplifikate mit Streptavidin beschichteten Dip-Sticks detektieren und die Varianten anhand ihres Laufverhaltens zuordnen. Die Varianten-Analyse mit RAY dauert nur eineinhalb Stunden, doch gilt auch hier: Die Methode erkennt nur bekannte Mutationen, wie zum Beispiel N501Y.
... oder Massenspektrometer
Einen Massenspektrometrie-basierten Varianten-Test des amerikanischen Herstellers Agena Bioscience hat der molekularbiologische Servicedienstleister Seq-It aus Kaiserslautern weiterentwickelt. Bei diesem sogenannten MassArray-System wird ein Panel, das fünf Varianten enthält, anhand von zwanzig genetischen Markern analysiert. Das Panel wird in 96- oder 384-Well-Platten mit den entsprechenden Primern überprüft. Dazu wird zunächst die RNA der Proben extrahiert. Anschließend amplifiziert man die relevanten Sequenzabschnitte mithilfe der Primer in einer One-Step-RT-PCR im Multiplexing-Format. Die entstandenen cDNAs werden danach in Gegenwart von Masse-modifizierten Terminator-Nukleotiden (ddNTPs) um genau ein Nukleotid (die SNP-Position) verlängert und auf einen sogenannten Matrix-precoated Spectro-Chip aufgetragen. Von diesem gelangen sie schließlich in ein Massenspektrometer, das die bei der Extension eingebauten ddNTPs analysiert und den jeweiligen SNPs beziehungsweise Varianten zuordnet.
Auch auf Protein-Ebene könnte die Massenspektrometrie bei der Suche nach Varianten weiterhelfen. Forscher um Kevin M. Downard von der University of New South Wales in Sydney, die sich schon lange mit SNPs in Influenzaviren befassen, übertrugen die mit diesen gewonnenen Techniken auf SARS-CoV-2 (ACS Infect. Dis. 6(12): 3269-76).
Der Proteotyping-Ansatz der Australier geht von kultivierten Viren oder von Abstrichen aus. Nach einer Polyethylenglycol-Fällung werden die Viren rekonstituiert und in einem Puffer mit Ultraschall behandelt, der für den anschließenden Verdau mit Trypsin versetzt wird. Danach wird die Probe im MALDI-Massenspektrometer analysiert. SARS-CoV-2 verrät sich in diesem durch einige Signatur-Peptide, die aus seinen häufigsten Proteinen S, E, M und N entstehen.
Ob die Australier ihr System tatsächlich zur Detektion von Varianten einsetzen können, ist aber noch offen. So ist zum Beispiel die prominente Aminosäure-Position N501 nicht unter den neun detektierbaren Peptiden, dafür aber K417, die in B.1.351 (K417N) sowie P.1 (K417T) mutiert ist.