Einzelzell-Analyse
Methoden-Special: Exakt verortete Transkripte
Karin Hollricher
(08.04.2021) In den letzten zehn Jahren machte die Einzelzell-Analyse erstaunliche Fortschritte. Ihr neuester Coup
ist die räumlich aufgelöste Transkriptomik, die Nature Methods zur Methode des Jahres 2020 kürte. Mit immer raffinierteren Techniken versuchen Forscher, Transkripte genau zu lokalisieren.
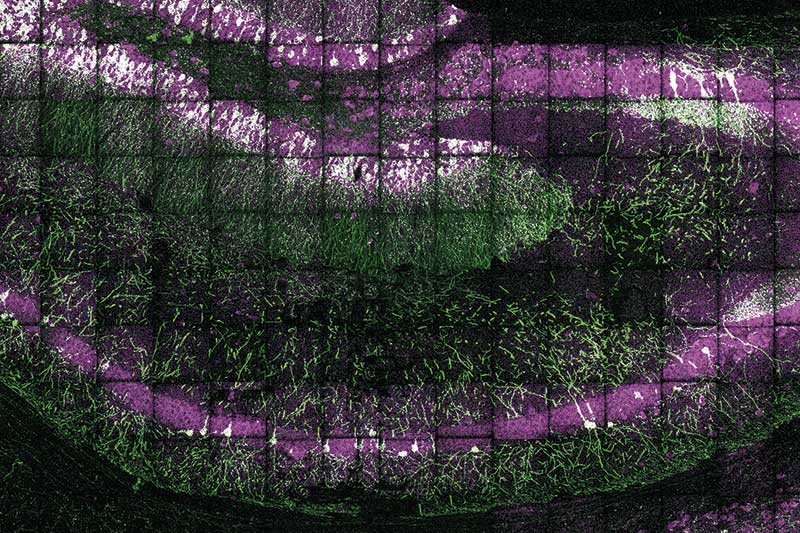
Die Einzelzell-Sequenzierung (scRNA-seq) ist fast schon wieder von gestern. Mit der ortsaufgelösten Transkriptomik (Spatially Resolved Transcriptomics) können Forscher inzwischen tausende Transkripte in Zellen lokalisieren und mikroskopisch abbilden, mitunter sogar subzellulär. Die räumliche Platzierung der genetischen Aktivität im histologischen Kontext und gleichzeitige Quantifizierung der Transkripte liefert entscheidende biologische Informationen – etwa über die Entwicklung von Zellen, Organen und Organismen, über Zellidentitäten, deren Funktionen sowie über interzelluläre Kommunikation.
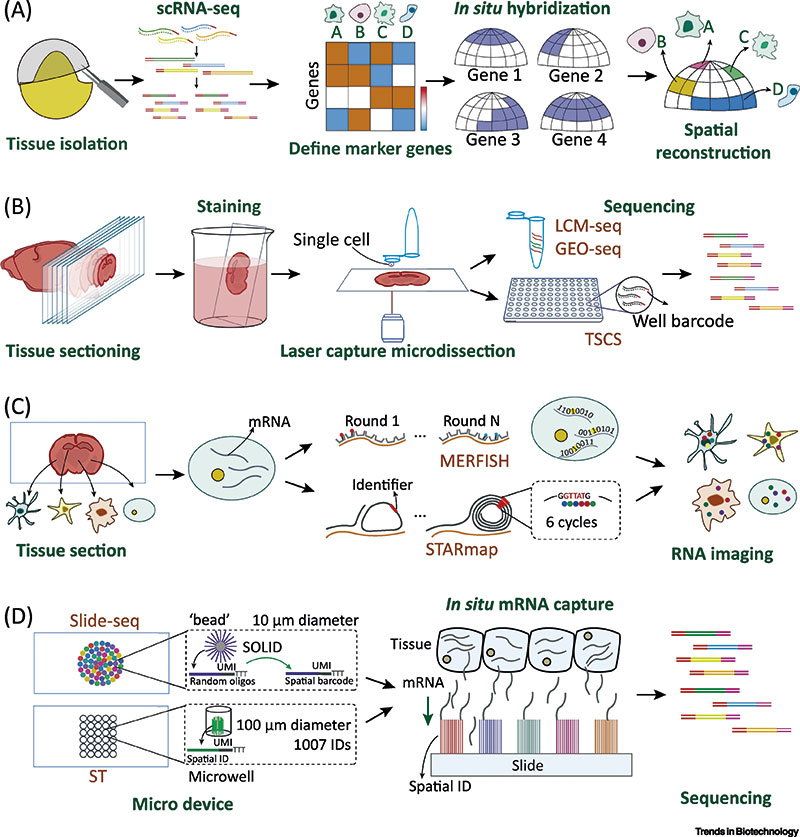
Wie gelingt es aber, einzelne Transkripte mit einem konfokalen Mikroskop zu lokalisieren, dessen optische Auflösung dafür bei Weitem nicht ausreicht? Sämtliche Technologien für die ortsaufgelöste Transkriptomik lassen sich im Wesentlichen in zwei Gruppen einteilen: Fluoreszenz-in-situ-Hybridisierung(FISH)-Methoden für RNA-Moleküle, deren Sequenz man bereits kennt; oder ungezielte (untargeted) Hochdurchsatz-RNA-Sequenzierungs-Techniken.
Da fast immer ein konfokales Mikroskop im Spiel ist, ist die Auflösung der RNA-Lokalisierung physikalisch auf die halbe Wellenlänge des verwendeten Lichts begrenzt. FISH-basierte Technologien erlauben eine zelluläre, meist sogar subzelluläre Darstellung der RNAs. Kombiniert man sie mit der Expansionsmikroskopie, bei der das Gewebe vor der Untersuchung mit Polymeren aufgebläht wird, lässt sich die Auflösung weiter steigern. Mit den Vertretern der Hochdurchsatz-Sequenzierung erreicht man den Beschreibungen der Forscher zufolge eine „fast-zelluläre“ Auflösung.
Wie immer hat jede Technologie Vor- und Nachteile. Bevor man sie im eigenen Labor etabliert, sollte man sich daher Gedanken über die wichtigsten Kriterien machen: Wie viele und welche Gene will ich nachweisen? Wie viele Zellen will ich analysieren. Auch die Kosten können durchaus eine Rolle spielen.
„Will man die Transkription weniger Gene in vielen Zellen verfolgen, benutzt man FISH-Technologien“, erklärt Evan Macosko vom Broad Institute in Cambridge (USA), einer der Hochburgen der Technologie-Entwicklung. „Will man aber die Aktivität tausender Gene beobachten, setzt man Sequenz-basierte Methoden ein, wie Slide-Seq.“
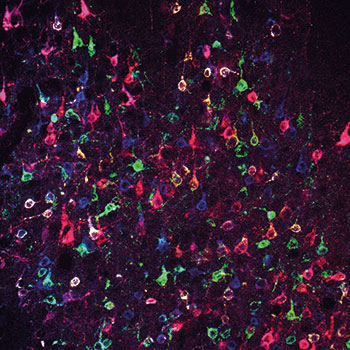
Konkret: Hat man es mit höchstens zehn Genen in maximal 100 Zellen zu tun, ist man mit seqFISH gut bedient. Um mehr Gene in mehr Zellen zu analysieren, ist MERFISH (Multiplexed Error-Robust Fluorescence In Situ Hybridization) in seinen bisher sechs Varianten, seqFISH+ oder FISSEQ (Fluorescent In Situ Sequencing) zu empfehlen. Den aktuell höchsten Durchsatz versprechen Slide-seq und HDST (High-Density Spatial Transcriptomics). Damit lassen sich 10.000 und mehr mRNAs in 10.000 oder 100.000 Zellen identifizieren und lokalisieren.
Schauen wir uns an einigen Beispielen an, wie die Technologien prinzipiell funktionieren. Zunächst die FISH-Vertreter. Um einzelne RNA-Moleküle durch In-situ-Hybridisierung mikroskopisch darzustellen, reicht ein Oligonukleotid (Oligo) mit einem daran gebundenen Fluorophor als Probe nicht aus. Man braucht ein stärkeres Signal. Bei ersten Versuchen verwendete man einfach mehrere, entlang der gesamten RNA bindende Oligos, die mit jeweils mehreren Farbstoffen markiert waren. Standard der smFISH (Single-molecule Fluorescence In Situ Hybridization) getauften Technologie waren fünf Proben mit jeweils fünf Fluorophoren. Obwohl die Oligos 50 Basenpaare lang waren, kamen sich die Fluorophore gegenseitig in die Quere – sowohl sterisch wie auch durch gegenseitige Löschung der Fluoreszenz (self-quenching).
Verstärktes FISH-Signal
Um das FISH-Signal zu amplifizieren, entwickelte man zwei prinzipiell verschiedene Varianten. Bei RNAscope dient ein längeres RNA-spezifisches Oligo als Basis für markierte Oligos, die man in einer zweiten Runde anhybridisiert. Die seqFISH-Technologie verwendet mehrere, kürzere Proben mit nur jeweils einem Fluorophor, die sequenziell hybridisiert werden (Nat. Methods 5: 877-9).
smHCR (Single-molecule Hybridization Chain Reaction) ist eine Sandwich-Technologie und Fortentwicklung von seqFISH. Hier werden an jedes Sequenz-spezifische Oligo gleich mehrere Fluorophor-Träger angedockt (Development 143: 2862-7). Limitierend ist die Anzahl der Fluorophore: Ihre Abstrahlung muss sich ausreichend unterscheiden, um sie mit dem Mikroskop auseinanderhalten zu können. Gleichzeitig kann man nur mit vier bis fünf Farbstoffen arbeiten.
Multiplex-Werkzeuge entstanden erst mit der Einführung von Farbcodes. Dabei wird jede RNA-Spezies nicht mit einer einzigen Fluoreszenzfarbe identifiziert, sondern durch eine Farbkombination. Die hierfür nötigen längeren Proben sind im mittleren Abschnitt komplementär zu individuellen RNAs. Die überhängenden Flanken enthalten hingegen mehrere unterschiedliche Sequenzen, die verschiedenfarbig markierte Auslese-Oligos binden können. Zunächst werden alle Proben an ihre spezifischen mRNAs anhybridisiert (mehrere Proben pro RNA-Molekül) und danach Runde für Runde jeweils ein Set farbmarkierter Auslese-Oligos getestet.
Binärer Code
Angenommen eine RNA zeigt in den Hybridisierungs-Runden mit drei verschiedenfarbig markierten Auslese-Oligos den Farbcode „Rot-keinSignal-keinSignal“, weil nur ein Auslese-Oligo an das RNA-spezifische Oligo bindet. Eine andere hybridisiert hingegen mit zwei Oligotypen aus denen der Farbcode „Rot-kein Signal-Grün“ resultiert. Formal entsteht hierdurch ein binärer Code für jeden Farbkanal: RNA, die in diesem Beispiel nur mit rot-markierten Oligos gelabelt wird, erhält nach drei Runden den Code 100, während RNA, die mit Rot und Grün markiert wurde, den Code 101 bekommt. Mit drei Farben lassen sich auf diese Weise 23 = 8 Farbkombinationen erreichen. Mit einem 16-stelligen Farbcode könnte man mehr als 65.000 individuelle Transkripte identifizieren. Im echten Laborleben beschränken sich die Forscher aber eher auf fünf- oder sechsstellige Barcodes (Science 348 (6233): aaa6090).
In der Abkürzung MERFISH steht „ER“ für Error-Robust: Die Auslese-Oligos sind so gewählt, dass sich die binären Codes für individuelle RNA-Spezies an mindestens zwei Positionen voneinander unterscheiden. In der jüngsten Version, MERFISH6, wird dieses Prinzip mit einer drastischen Amplifikation der Auslese-Oligos kombiniert. Die Signalstärke wurde um den Faktor 30 gesteigert. Das hat den Vorteil, dass man weniger Proben an jede einzelne RNA binden muss und somit auch kürzere Moleküle detektieren kann. Über 10.000 verschiedene Transkripte konnten damit in einer Zelle identifiziert werden (Sci. Rep. 9: 7721).
seqFISH+ funktioniert prinzipiell ähnlich wie MERFISH und ist ebenso leistungsstark (Nature 568: 235-9).
FISH und verwandte Technologien kann man für die räumlich aufgelöste Transkriptomik nur verwenden, wenn man die Sequenzen der zu analysierenden RNAs bereits kennt. Ist dies nicht der Fall, oder will man möglichst alle RNA-Spezies einer Zelle erwischen, muss man eines der Verfahren mit Hochdurchsatz-Sequenzierung einsetzen, wie beispielsweise Slide-Seq (Nat. Biotechnol. 39: 313-9).
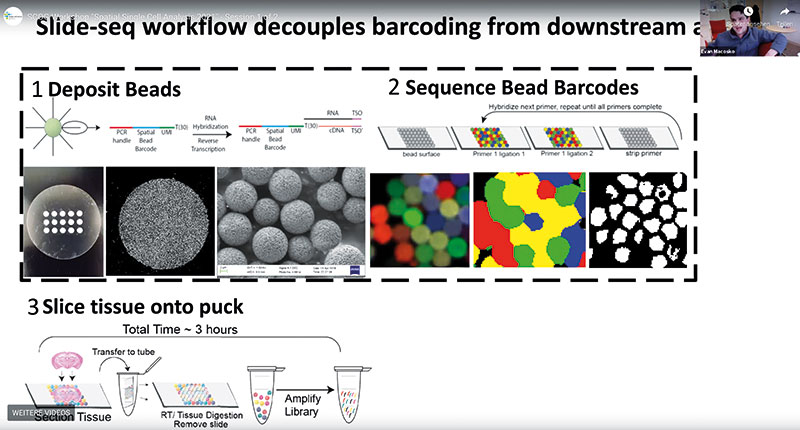
Für Slide-Seq verankert man kleine Kügelchen (Beads) so auf einem Glasplättchen, dass sie dicht an dicht liegen und nur eine Lage bilden. Die Beads sind mit Oligos bestückt, die wie bei der Sequenzierung der Transkripte einzelner, isolierter Zellen aus vier Komponenten bestehen: Einem polyA-Ende, um mRNAs zu binden (nicht-codierende RNAs fallen also durch das Raster); einem Primer, um die cDNA-Synthese von der mRNA zu starten; ID-Sequenzen, die alle an ein Kügelchen gebundenen Oligos genau diesem zuordnen, sowie UMI-Sequenzen, die jedes der an ein Kügelchen gebundenen Oligos individuell markieren. Damit lassen sich sämtliche cDNAs, die man von jeweils einem Oligo gewinnt, exakt der daran gebundenen mRNA zuordnen.
Genaue Positionierung
Die Position jedes Kügelchens wird anhand seines Barcodes bestimmt – dann kann das Experiment beginnen. Auf die Bead-Schicht platziert man den Dünnschnitt eines gefrorenen Gewebes. „Man muss das Gewebe auf den Beads nicht fixieren, denn sie sind so unglaublich klebrig, dass sie mit den Zellen förmlich verschmelzen“, erklärt Macosko. „Deshalb kann man das Glasplättchen samt Gewebe mit Hybridisierungspuffer inkubieren, ohne dass die Nukleinsäuren verschwimmen, bevor sie gebunden haben.“
Anschließend folgt die cDNA-Synthese auf dem Glasplättchen in einem Eppendorfgefäß. Jedes Kügelchen hat einen Durchmesser von zehn Mikrometern, deckt also in etwa die Fläche einer Zelle ab. Bestenfalls binden also alle mRNAs von nur einer Zelle an ein einziges Kügelchen. Man kann aber nicht ausschließen, dass sich daran auch Transkripte einer benachbarten Zelle anlagern. Für ihre Slide-SeqV2-Version verbesserten die Forscher die Bestückung der Kügelchen mit Oligos und die Synthese des zweiten DNA-Strangs. Ein Experiment dauere jetzt nur noch maximal zwei Tage, freut sich Macosko.
Nach ähnlichen Prinzipien funktionieren auch die Plattform Visium der Firma 10xGenomics und das High-Definition Spatial Transcriptomics(HDST)-Verfahren von Fredrik Salmén, Patrik Ståhl, Joakim Lundeberg und Kollegen vom KTH Royal Institute of Technology in Stockholm (Nat. Protoc. 13: 2501-34). Die Stockholmer waren tatsächlich die Ersten, die eine Methode zur ortsaufgelösten Darstellung der Transkriptome einzelner Zellen entwickelten und den Begriff Spatial Transcriptomics in die Welt setzten (Science 353: 78-82). Selbst an Pflanzen, deren Zellen dank einer Zellwand deutlich stabiler sind als diejenigen von Tieren, lassen sich die Transkriptome einzelner Zellen ortsaufgelöst darstellen (Nat. Protoc. 13: 2425-46).
UV-empfindlicher Linker
Etwas anders funktioniert der „GeoMx Digital Spatial Profiler“ der Firma NanoString. Hier verschmelzen Hybridisierung und ungezielte Sequenzierung miteinander. Dabei setzt man markierte Oligos ein, die einen durch UV-Licht schneidbaren Linker enthalten. Auf einer Seite des Linkers sitzt eine zur RNA komplementäre Sequenz, auf der anderen befindet sich ein Code zur Identifizierung des Konstrukts. Zu jeder spezifischen RNA-Sequenz gibt es also einen spezifischen ID-Code.
Der Gewebeschnitt wird mit den Oligos hybridisiert, anschließend wäscht man nichtgebundene Moleküle ab. Danach wählt man unter dem Mikroskop eine interessante Region aus und belichtet sie. Der Ausschnitt kann sich auf eine Zelle beschränken, aber auch deutlich größer sein. Durch die Belichtung werden die ID-Codes freigesetzt und danach eingesammelt. Eine cDNA-Synthese ist nicht nötig, denn man kann über die Codes herausfinden, von welchen RNAs sie stammen. Der „GeoMx Whole Transcriptome Atlas“ von NanoString enthält Oligos für mehr als 18.000 Gene. Die Technik funktioniert auch mit Formalin fixierten und in Paraffin eingebetten Proben.
Mit Licht arbeitet auch die ZipSeq-Methode, die Forscher der University of California in San Francisco entwickelten (Nat. Methods 17: 833-43). ZipSeq kennzeichnet die zu untersuchenden Zellen quasi nach Bedarf. Dafür werden sie auf ihrer Oberfläche mit Lipiden oder Antikörpern markiert, die jeweils ein Oligo mit einem Primer für die cDNA-Synthese enthalten. In einer zweiten Hybridisierung werden Barcode-Oligos mit poly(A)-Sequenzen für das Einsammeln von mRNAs angeheftet. Dies ist aber erst möglich, wenn man durch UV-Licht eine Blockade auf dem ersten Oligo entfernt hat. Über das Licht wählt man die Zellen aus, die man analysieren will, anschließend werden sie vereinzelt, um Transkriptome aus ihnen zu generieren.
Bei diesem Verfahren verschmilzt die räumliche Einzelzell-Transkriptomik mit Tropfen-basierten scRNA-Methoden. Man kann viele Zellen oder Regionen sukzessive belichten, denn die daraus generierten Sequenzen lassen sich später über den Code eindeutig zuordnen. Die Auflösung ist zurzeit aber noch nicht gut genug, um einzelne Zellen isoliert zu untersuchen. Doch die Forscher glauben, dass ZipSeq das Potenzial zur echten Einzelzell-Transkriptomik hat. Mit dieser ersten Variante, die sowohl in vitro wie ex vivo funktioniert, überprüften sie die Transkriptome von verwundetem Gewebe, von Tumoren und von Lymphknoten. In allen Fällen, so schreiben sie, hätten sie neue Expressions-Muster entdeckt und mit den jeweiligen histologischen Strukturen assoziieren können.
Hochdurchsatz mit Nanobällen
Eine weitere gängige Technik ist die Sequenzierung der lokalen Transkriptome mittels DNA-Nanobällen. Die Nanoball-Sequenzierung wurde als Hochdurchsatz-Verfahren vor gut zehn Jahren für die Genomanalyse eingeführt. Die RNA wird über eine cDNA-Synthese nach dem Rolling-Circle-Prinzip direkt auf den Zellen amplifiziert, wobei sich die cDNAs wie ein Wollknäuel aufwickeln. Jedes Amplikon enthält viele Kopien einer einzelnen RNA.
Diese Methode wurde von George Churchs Arbeitsgruppe in Boston weiterentwickelt und FISSEQ getauft (Nat. Protoc. 10: 442-58). Vor gerade mal zwei Monaten folgte dann ein Knaller: Church und 40 weitere Autoren (die meisten von Instituten in Boston und Cambridge) beschrieben, wie sie FISSEQ optimiert und in Kombination mit der Expansionsmikroskopie benutzt hatten (ExSeq), um individuelle RNA-Moleküle mit einer Auflösung von vermutlich einigen Hundert Nanometern darzustellen (Science 371 (481): eaax2656). Leider schreiben die Autoren in ihrem Paper immer nur „Nanoscale“ und machen keine exakten Angaben zu den Dimensionen. Jedenfalls konnten sie mit ihrer Methode die normalerweise sehr dicht gepackten Transkripte räumlich voneinander trennen und einzeln sequenzieren.
Die Experimente von Church und Co. beschränkten sich aber nicht nur auf ein Beispiel oder einen Organismus. Die Gruppe testete die Methode umfassend an verschiedenen Untersuchungsobjekten. Dazu zählten Gehirne von Mäusen, Caenorhabditis elegans, Embryonen von Drosophila melanogaster, HeLa-Zellkulturen und Brustkrebs-Biopsien.
Mit der ungezielten Sequenzierung identifizierten die Forscher im Mausgehirn neben reifen mRNAs auch Transkripte mit Introns sowie lange nicht-codierende RNAs. In dem aufgeblähten Gewebe sahen sie, dass bestimmte RNAs sich eher in den Dornfortsätzen von Neuronen, andere eher in benachbarten Dendriten aufhalten. „Thus ExSeq enables highly multiplexed mapping of RNAs, from nanoscale to system scale“, resümieren die Autoren in ihrem jüngst erschienenen Artikel (Science 371 (6528): eaax2656 ).
Aufblähen und sequenzieren
Allerdings waren sie nicht die Ersten, die Expansionsmikroskopie benutzten, um die Lokalisierung der Transkripte einfacher zu machen. Forscher vom CalTech in Pasadena und der Universität Harvard hatten schon 2019 mit seqFISH+ vorgemacht, wie sich Expansionsmikroskopie sinnvoll mit der Transkriptom-Analyse kombinieren lässt. Sie wiesen damit 24.000 RNA-Spezies in einer einzelnen Zelle nach (Nature 568: 235-9).
Mit DNA-Nanobällen experimentieren auch Forscher in China – kein Wunder, denn das Beijing Genomics Institute kaufte 2013 die Firma Complete Genomics, welche die Technologie entwickelt hatte. In einem in bioRxiv im Januar abgelegten Manuskript berichten sie über Stereo-Seq (doi: 10.1101/2021.01.17.427004). Die Forschenden expandierten den Barcode-Pool auf 425 Varianten(!), bestückten damit die an Nanobällen fixierten Primer und positionierten sie auf einem mit winzigen Löchern strukturierten Silizium-Chip. Jeder Ball hat einen Durchmesser von 220 Nanometer, der Abstand zwischen den Zentren zweier Bälle beträgt je nach Chip-Design 500 oder 715 Nanometer. Die Chinesen sequenzierten die Barcodes der fixierten Bälle, legten das zu untersuchende Gewebe darauf ab, ließen cDNA synthetisieren, sequenzierten diese und ordneten die Sequenzen über die Barcodes einer Position auf dem Chip zu.
In ihrem Artikel schreiben die chinesischen Forscher, sie hätten Chips von bis zu 200 Quadratmillimetern hergestellt und damit die Transkriptome der Zellen eines halben Mausgehirns analysiert. Unglaublich, wie rasant die Entwicklung voranschreitet. Womöglich lassen sich in wenigen Jahren alle Omiken an einzelnen, lokalisierten Zellen am lebenden Objekt durchführen und die Daten miteinander kombinieren.
Mit der Beschreibung der Technologien, die für die Spatially Resolved Transcriptomics eingesetzt werden, könnte man mehrere Laborjournal-Ausgaben füllen. Detailliertere Informationen, die über diesen Überblick hinausgehen, finden Sie in einem fast druckfrischen Review in Trends in Biotechnology (39 (1): 43-58) sowie auf der tollen Webseite (www.singlecell.de), die von Fabian Theis (Helmholtz Zentrum München), Nikolaus Rajewsky (MDC Berlin) und Jörn Walter (Universität des Saarlandes, Saarbrücken) ins Leben gerufen wurde. Unter dem Datum 10. Februar findet man den virtuellen Workshop „Spatial Single Cell Analysis“ mit Vorträgen führender Entwickler verschiedener Technologien. Alle 14 Tage wird ein Online-Vortrag angeboten. Außerdem ist die Webseite vollgepackt mit Informationen zu verschiedenen Single-Cell-Omiken.
Technologie ist aber das eine, Biologie das andere. Was also liefern die vielen Transkriptom-Daten an neuen Erkenntnissen? Die Genexpression der einzelnen Zelle definiert ihren Status, ihre Eigenschaften, ihre Identität. Darum verwendete man die Daten zunächst zur Zell-Typologie und entdeckte bisher unbekannte Zelltypen beispielsweise in der Lunge, den Gastrula-Stadien der Maus und im Gehirn. Bereits bekannte Zellen ortete man auch an unvermuteten Stellen. Auf diese Weise entstanden viele Zell-Atlanten, etwa von verschiedenen Organen mehrerer Modellorganismen, von Organen des Menschen und auch von Organoiden sowie von hämatopoetischen Stammzellnischen und soliden Tumoren.
Besonders die Entwicklungsbiologie dürfte von der Spatially Resolved Transcriptomics profitieren, denn die individuelle Genexpression steuert die Zukunft jeder einzelnen Zelle und dies beeinflusst auch die Entwicklung der Nachbarschaft. Die Lokalisierung wichtiger Kontrollgene und deren Transkripte ist also essenziell, um zu verstehen, welchen Weg eine Zelle bei der Entwicklung von der befruchteten Eizelle zum erwachsenen Organismus einschlagen wird. Das ist eine gleichzeitig sehr verlockende wie auch, der großen Zahlen wegen, extrem komplexe Aufgabe. Der Traum von einem Ontogenie-Atlas, der die Geschichte, den aktiven Status und die Zukunft der Zellen eines Organismus beschreibt, könnte mithilfe der ortsaufgelösten Transkriptomik aber tatsächlich wahr werden.