Molekulare Verkupplungs-Strategien
Neue Klonierungstechniken
Andrea Pitzschke
Die Klonierung von DNA-Fragmenten in Vektoren und Plasmide ist eine der Standardtechniken in molekularbiologischen Laboren. Neue Klonierungs-Verfahren erleichtern die Arbeit oder ermöglichen den schnellen und kostengünstigen Zusammenbau einzelner DNA-Bausteine zu größeren Einheiten.
Die Wege zu sauberen Proteinen, Lokalisationsstudien, Aktivitäts-Assays oder transgenen Organismen sind lang und steinig. Sie beginnen aber praktisch immer mit einer Klonierung. Die dabei verwendeten Strategien unterscheiden sich teils gravierend, ihre Ziele sind aber immer gleich: Die Klonierung sollte fehlerfrei verlaufen sowie mit wenig Kosten und noch weniger Aufwand verbunden sein. Dass regelmäßig neue Klonierungsmethoden zu den etablierten Verfahren hinzukommen, zeigt, dass es noch Luft nach oben gibt. Drei interessante neue Klonierungstechniken stellt dieses Special vor.
Klonierung bedeutet im Grunde nichts anderes, als DNA-Stücke aneinanderzukleben. Das lässt sich auf drei verschiedene Arten bewerkstelligen: Bei der direktionalen Klonierung schneidet man Insert und Vektor mit zwei unterschiedlichen Restriktionsenzymen und verkittet die durch einzelsträngige Überhänge entstandenen klebrigen Enden mit einer Ligase.
Die zweite Variante, die T/A-Klonierung, nutzt die Eigenart der DNA-Polymerase aus, einen A-Überhang an PCR-Produkte anzuhängen, der zum T-Überhang des Klonierungsvektors passt (zum Beispiel pGemT).
Zufällige Einbaurichtung
Die dritte Technik ist die sogenannte Blunt-End-Klonierung, bei der sowohl Insert als auch Klonierungsvektor glatte Enden aufweisen. In diesem Fall ist die Einbaurichtung dem Zufall überlassen. Damit das Insert überhaupt integriert wird und der linearisierte Vektor sich nicht einfach in den eigenen Schwanz beißt und rezirkularisiert, dephosphoryliert man in der Regel seine Enden. Die meisten Klonierungen laufen in vitro ab: Die Bausteine und Enzyme werden zusammengegeben und erst hinterher in einen Organismus (meist E. coli) verfrachtet.
Will man einfach nur ein Fragment in einen Vektor klonieren, ist die Sache ziemlich simpel. Die Klonierungs-Wünsche vieler Forscher sehen aber meist etwas komplizierter aus: Einige wollen auch große und/oder mehrere Fragmente gleichzeitig in den Klonierungsvektor einbauen. Andere möchten Zielgene zwischen verschiedenen Vektoren hin und her jonglieren. Das setzt universelle, kompatible Ein- und Ausbaustellen voraus. Vom rekombinanten Protein X mit His-Tag aus E. coli ist es dann nicht mehr weit bis zur YFP-getaggten Variante für Lokalisationstudien in transgenen Tabakblättern.
Der nächste hat keine Lust darauf, jede per Zufall gepickte transformierte Zelle arbeits- und kostenaufwändig zu sequenzieren, und braucht dazu ein Klonierungssystem, mit dem er schon vorab vielversprechende Kandidaten erkennen kann. Wer schließlich ein rekombinantes Protein exprimieren will, benötigt ein Klonierungssystem, bei dem das Zielgen direkt als PCR-Produkt im Expressionvektor landet – und nicht erst in einen Standardvektor kloniert, dann sequenziert und anschließend in den Expressionsvektor subkloniert werden muss.
Direktional und klebrig
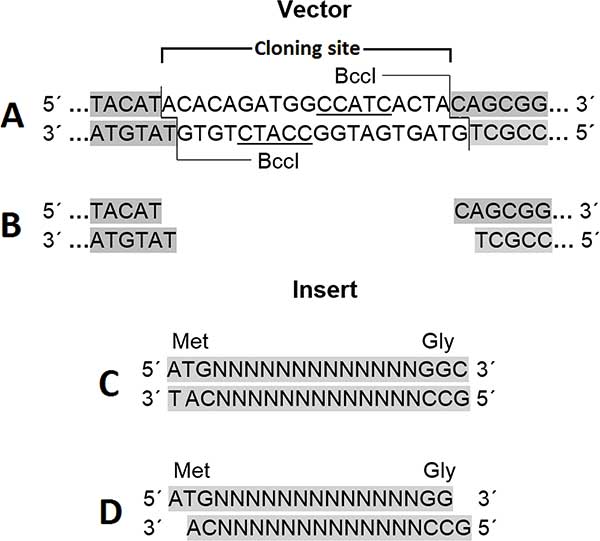
Einige dieser Wünsche erfüllt die TA-GC-Klonierung, die eine griechische Gruppe Ende letzten Jahres vorstellte (PLoS ONE 12(11): e0186568). Die TA-GC-Klonierung basiert auf A- oder G-Überhängen am Zielgen-Anfang beziehungsweise -Ende, die komplementär zu T- oder C-Überhängen am linearisierten Vektor sind. Sie ist ein typisches Beispiel für eine direktionale Klonierung mit klebrigen Enden (Sticky Ends).
Und wie kommen A und G an die Enden des DNA-Fragments oder Gens? Zunächst wird das Gen mit passenden Primern amplifiziert, wobei der Forward-Primer ein ATG (Startcodon) beisteuert und der Reverse-Primer im Amplikon ein GGC (Glycin) hinterlässt. Nach der Aufreinigung wird das PCR-Produkt mit T4-DNA-Polymerase in Gegenwart von dATP und dTGP inkubiert. Aufgrund der 3´-5´-Exonuklease-Aktivität frisst sich die T4-Polymerase von den Enden her in das PCR-Produkt hinein, bis sie auf ein A oder G stößt. Bei der TA-GC-Klonierung passiert dies schon beim ersten Nukleotid-Happen. Vom Antisense-Strang-Ende geht hierdurch das 3´-T verloren (die Partnerbase zu ATG), vom Sense-Strang das 3´-C. Es verbleibt ein PCR-Produkt mit 5´-Überhang (A am Anfang, G am Ende).
3‘-Überhang durch BccI
Der Zielvektor wird mit einem Trick mit den passenden klebrigen Enden ausgestattet: Man verdaut ihn mit dem TypII-Restriktionsenzym BccI, das den Sense- beziehungsweise Antisense-Strang fünf beziehungsweise vier Basen hinter dem Erkennungsmotiv CCATC schneidet. BccI hinterlässt dadurch einen 3´-Überhang. Stehen sich zwei BccI-Motive benachbart gegenüber, so schneidet das Enzym einmal davor und einmal dahinter, wodurch das Motiv herausgeschnitten wird. Übrig bleibt der linearisierte Vektor mit klebrigen Enden, die zu den Überhängen des vorbereiteten Inserts passen. Von hier geht es routinemäßig weiter mit Ligation, Transformation und Selektion.
Was bringt dieser Aufwand verglichen mit den bisherigen Strategien? Das Wunschgen kann direkt nach der Amplifikation in einen Expressionsvektor eingebaut werden, ohne einen Zwischenstopp in einem Klonierungs-/Sequenzierungsvektor einlegen zu müssen. Interne Restriktionsstellen, die dazu führen, dass ein Zielgen im klassischen bidirektionalen Verfahren zerhackt wird, sind kein Problem. Zudem muss man die genaue Sequenz des Zielgens nicht kennen – für das Primer-Design reicht die Sequenz der Enden. Ist der Zielvektor ein Expressionsvektor, so lassen sich zudem die vielversprechendsten Transformanten vorab identifizieren.
Das Verfahren sollte sich auch mit Zufallsmutations-Strategien kombinieren lassen. Bei einer absichtlich fehlerhaften PCR entstehen verschiedene Varianten des Zielgens. Solange diese ihr ATG beziehungsweise GGC an den Enden behalten, kann man sie mit der TA-GC-Klonierung exprimieren. Das Zielgen ist mit einer einzigen PCR und T4-DNA-Polymerase-Behandlung schnell hergestellt und in einen BccI-Vektor kloniert – vorausgesetzt, die entsprechenden BccI-Vektoren stehen parat. Das griechische Team hat bisher nur einen pET-BccI-Klonierungsvektor konstruiert, bei dem die klassische multiple Klonierungsstelle durch eine Tandem-BccI-Stelle ausgetauscht wurde. Getestet hat die Gruppe die Methode an drei Demo-Genen, die bis zu zwei Kilobasenpaare lang waren.
Lieber ohne Kanamycin-Marker
Dass etwa 80 Prozent der Kolonien nach der Transformation chemisch kompetenter Zellen (den Stamm verraten die Autoren nicht) das Konstrukt enthielten und 98 Prozent davon tatsächlich (nach IPTG-Induktion) exprimierten, klingt gut. Wer auf das System umsteigen will, sollte jedoch um Vektoren mit Kanamycin-Selektionsmarker einen Bogen machen. Es sei denn, man schreckt nicht vor der kniffligen Aufgabe zurück, die darin liegenden BccI-Motive zu mutieren, ohne die Resistenzfunktion zu zerstören. Mit der TA/GC-Methode kommt man hier jedenfalls nicht weiter.
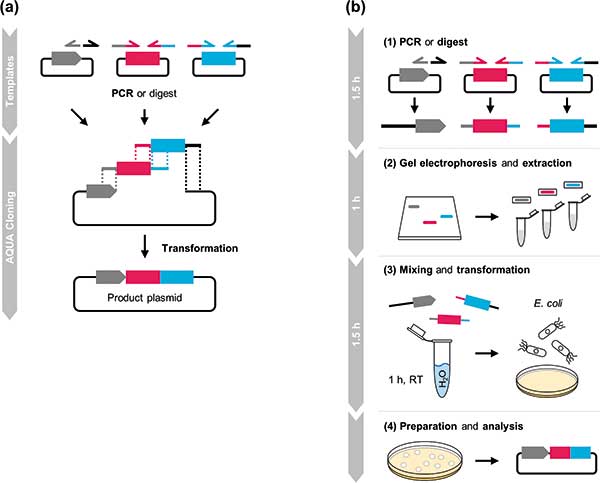
Das Abtauchen in die Klonierungs-Literatur förderte mit der Advanced Quick Assembly- kurz AQUA-Klonierung jedoch eine weitere neue Klonierungstechnologie zutage, die nicht nur Mutations-, sondern auch (multiple) Insertions- und Deletions-Aufgaben meistert. Entwickelt wurde sie von Matias Zurbriggens Gruppe, damals noch am Freiburger BIOSS Centre for Biological Signalling Studies (PLoS ONE 10(9): e0137652).
Einfaches Zusammenfügen
Die AQUA-Klonierung gehört zur Kategorie der Assemblierungs-Techniken, die insbesondere in der synthetischen Biologie dazu genutzt werden, lineare DNA-Fragmente aneinander zu hängen und in zirkuläre Plasmide zu überführen. Nach dem, was im Abstrakt des Papers der Zurbriggen-Gruppe zu lesen ist, braucht man für sie weder Kits, Enzyme oder spezielle Reaganzien. Bei der AQUA-Klonierung verlängert man die Enden von DNA-Fragmenten, die miteinander verknüpft werden sollen, zunächst mittels PCR um 16 bis 32 homologe Basenpaare. Anschließend schleust man die DNA-Fragmente in E. coli-Zellen ein. Treffen die homologen Abschnitte in der Zelle aufeinander, schmiegen sie sich aneinander und das DNA-Reparatursystem der Zelle verknüpft sie in vivo.
Im Gegensatz zu klassischen Ligationen werden die Enden bei der AQUA-Klonierung nahtlos ohne „Narben“ miteinander verbunden (seamlees cloning). Zwar gibt es auch diverse kommerzielle Kits für den narbenfreien DNA-Zusammenbau – diese funktionieren aber nur in vitro und sind nur etwas für Gruppen mit dicken Brieftaschen. Interessanterweise hat sich die Hypothese der Gruppe: „Je länger die homologe Region, desto effizienter der Zusammenbau“, nicht bestätigt.
Da auch hier, wie bei allen Experimenten, nicht immer alles glatt verläuft, enthält das Paper einen Troubleshooting Guide. Akribisch haben Zurbriggens Mitabeiter verschiedene E. coli-Stämme getestet und alle als „qualifiziert“ für die AQUA-Klonierung bewertet. Darunter auch den Expressions-Stamm BL21. Das ist sehr praktisch, denn so verkürzt sich die Zeit vom ersten Pipettengriff bis zum geernteten rekombinanten Protein auf schlappe 24 Stunden. BL21 hat auch keine Probleme damit, mehrere DNA-Fragmente auf einmal zu verarbeiten. Das Team zeigt dies anhand der Ligation von mCherry in einen T7-Expressionsvektor. Die bewußt zerstörte Resistenzkassette des Vektors wurde durch die homologe Rekombination der Fragmente wieder zusammengeschweißt.
Die AQUA-Klonierung taugt aber auch für Mutations-Experimente. Ihr Prinzip – nicht aber der technische Ablauf – ähnelt dann der klassischen zielgerichteten In-vitro-Mutagenese (Site-directed Mutagenesis), für die jedoch Gelelektrophorese, Fragmentaufreinigung, Ligation und weitere Schritte nötig sind.
Schneller und einfacher geht es mit der AQUA-Klonierung: Zwei überlappende Primer mit zusätzlichen 32 Basenpaaren sowie der gewünschten Mutation binden an der entsprechenden Stelle des Zielgens und werden durch eine PCR verlängert. Die homologen Enden der entstandenen Amplikons finden nach der Transformation in E. coli zueinander und werden verknüpft.
Vielversprechend könnte die AQUA-Klonierung auch für Kombinations-Kunststücke wie das Mischen von Domänen sein. Angenommen ein langes Protein mit den Domänen A, B und C stammt aus einer Genfamilie mit 30 Vertretern. Gut möglich, dass Domäne A von Vertreter 28, Domäne B von Vertreter 14 und Domäne C von Vertreter 2 das perfekte Protein, etwa ein sehr aktives Enzym, ergibt. Mit der AQUA-Klonierung kann man einfach den Zufall walten lassen und alle Kombinations-Möglichkeiten durchspielen. Voraussetzung ist nur, dass die Domänenübergangs-Abschnitte genügend konserviert sind, um das Primer-Annealing zu gestatten.
Noch zu selten genutzt
Bisher stieß die AQUA-Klonierungs-Strategie aber auf eine überschaubare Resonanz – zumindest wurde das Original-Paper von Ende 2015 erst ein Dutzend mal zitiert. Die Vielfalt und Komplexität der publizierten Anwendungen legen aber nahe, dass die Methode noch viel Potenzial hat. So hat zum Beispiel Gerald Radziwills Gruppe von der Universität Freiburg mit ihr verschiedene Konstrukte zur Expression lichtaktivierbarer Kinasen hergestellt, die sie in menschlichen Zellkulturen einsetzte (Cellular Signalling 42: 176-83).
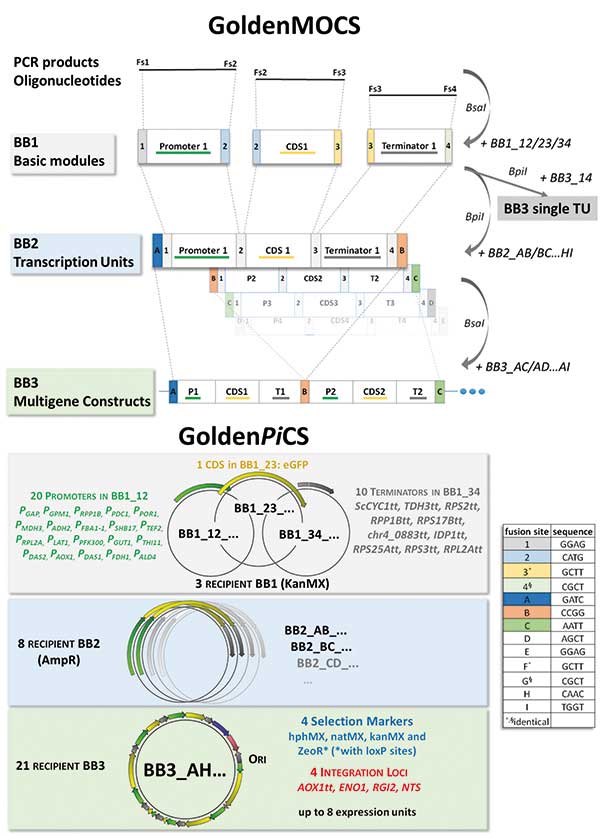
Nicht nur für Fans der Hefe Pichia pastoris dürfte das Golden Gate P. pastoris-Klonierungs-System (Golden PiCS) interessant sein, das die Gruppe von Hans Marx von der Universität für Bodenkultur Wien entwickelte (BMC Systems Biology 11:123). Mit Golden PiCS ist es möglich, das genetische Programm für komplette Stoffwechselwege innerhalb von neun Tagen in Pichia pastoris unterzubringen. Mit klassischen Verfahren dauert dies gut und gerne mehr als einen ganzen Monat. Mehrere Plasmide nacheinander einzuschleusen dauert ebenfalls lange und ist nicht besonders effizient – für jedes Plasmid benötigt man neue kompetente Zellen und muss diese anschließend auf den eingebauten Vektor absuchen.
Vormontierte Bausteine
Die Golden PiCS-Technik ist eine auf P. pastori zugeschnittene Variante des sogenannten Golden Gate-derived Multiple Organism Cloning Systems (Golden MOCS). Sie basiert auf dem Vormontieren kleinerer Bausteine zu Blöcken, die anschließend miteinander verknüpft werden. Hierzu tragen der Promotor, die kodierende Sequenz des Wunschgens (CDS) und der Transkriptons-Terminator an den Enden aufeinander abgestimmte Fusions-Stellen (Fusion sites, FS), die vier Basenpaare lang sind. Die FS am 3´-Ende des Promotors passt zum 5´-Ende der CDS. Das 3´-Ende der CDS passt wiederum zur 5´-FS des Terminators. Die Sequenzen der Fusionsstellen können zufällig sein – mit einer Ausnahme: Jeder CDS-Anfang und damit auch jedes Promoter-Ende muss das ATG-Startcodon tragen.
Mittels Overlap Extension-PCR wird so ein dreiteiliger Block aus Promotor, CDS und Terminator amplifiziert und verkittet. Dasselbe spielt man mit den Promotoren, CDS und Terminatoren der weiteren Proteine des Stoffwechselweges durch. Als Zwischenstufe erhält man für alle Proteine eines gewünschten Stoffwechselwegs die entsprechende Expressionskassette (Transcription Unit). Anschließend verknüpft man diese Units (bisher sind maximal acht möglich) zu einer langen Kette. Die Reihenfolge der Glieder definiert man vorher über die Sequenz der ersten und letzten Fusionsstelle jeder Unit. So passt das Terminator-Ende der ersten Transcription Unit zum Promotor-Start der zweiten Transcription Unit und so weiter. Die Kupplung erfolgt diesmal nicht über Overlap Extension-PCR (und umgeht so mögliche Lesefehler der Polymerase), sondern über eine Ligation. Wie bei der Golden Gate-Klonierung verwendet man TypII-Restriktionsenzyme (BpiI, BsaI), die außerhalb ihrer Erkennungssequenz schneiden, und kann hierdurch Verdau und Ligation als Ein-Topf-Reaktion durchführen.
Viele Werkzeuge vorhanden
Verblüffend, dass Pichia pastoris den mit der Golden PiCS hergestellten riesigen Plasmid-Brocken tatsächlich schluckt. Mit zwanzig Promotoren, zehn Transkriptions-Terminatoren und vier Resistenzmarkern ist die Golden PiCS-Werkzeugkiste des Wiener Forscherteams gut gefüllt. Je nach Wahl des Promotors kann man die Expression der einzelnen Gene des Stoffwechselwegs steuern und so das Tempo auf jeder Stufe anpassen.
Ob in vivo oder in vitro, für einfache Konstrukte oder komplexe Stoffwechselwege: Für nahezu alle molekularbiologischen Herausforderungen findet sich eine passende Klonierungsmethode. Und wer den Mut hat, neue Klonierungstechniken auszuprobieren, und die mögliche anfängliche Frustrationsphase überwindet, kann in vielen Fällen nicht nur Zeit, sondern auch bares Geld sparen.
Letzte Änderungen: 05.06.2018