Welche Technologie macht das Rennen? - Next Generation Protein Sequencing
Michael Bell
(13.05.2022) Noch dominieren Massenspektrometrie-gestützte Proteom-Verfahren die Protein-Sequenzierung.
Die Zukunft könnte jedoch der Sequenzierung auf Einzelmolekül-Ebene gehören. Ein heißer Kandidat hierfür ist die Protein-Sequenzierung mit Nanoporen.
Ein unscharfes schwarzes X auf weißem Grund – die Röntgenaufnahme Rosalind Franklins, mit deren Hilfe James Watson und Francis Crick 1953 die DNA-Doppelhelix entschlüsselten, kennt fast jeder. Damit war die räumliche Struktur der Erbsubstanz vollständig aufgeklärt, die vier Basen als Grundbausteine waren schon länger bekannt. In den Siebzigerjahren entwickelte der Biochemiker Frederick Sanger erstmals eine Methode, um die Basenabfolge in einem DNA-Strang exakt zu bestimmen. Die Sanger-Sequenzierung war für Jahrzehnte der Goldstandard und brachte dem Erfinder 1980 den Nobelpreis für Medizin oder Physiologie ein.
Knapp zwei Dekaden später eroberten Next-Generation-Sequencing (NGS)-Methoden den Markt. Sie ermöglichten Genetikern, Millionen von Basen in nie dagewesener Geschwindigkeit zu sequenzieren. Inzwischen werden ganze Genome in 24 Stunden ausgelesen.
Frühe Anfänge
Und wie sieht es mit der Sequenzierung von Proteinen aus? Tatsächlich reichen die Anfänge der Proteinforschung deutlich weiter zurück als die der DNA-Forschung. Schon Anfang des zwanzigsten Jahrhunderts kam die Theorie auf, dass Proteine aus langen Aminosäureketten bestehen. Zu diesem Zeitpunkt waren bereits 17 der 20 Protein-bildenden Aminosäuren bekannt. Die Proteine standen im Mittelpunkt des Interesses, weil man sie für die Träger der Erbinformation hielt – und nicht die kaum erforschte DNA. Es herrschte jedoch lange Zeit kein Konsens darüber, ob Aminosäuren allein für den Aufbau der komplexen Biomoleküle verantwortlich sind. Das änderte sich, als der Biochemiker Albert Chibnall von der Cambridge University einen ambitionierten Nachwuchsforscher anheuerte, um die Sequenz des Enzyms Insulin zu knacken. Sein Name: Frederick Sanger. Im Jahr 1955 sequenzierte Sanger mit Insulin erstmals ein Protein vollständig. Fast zwei Jahrzehnte vor den wegweisenden Arbeiten an DNA erhielt er 1958 seinen ersten Nobelpreis, was ihn zu einem von nur vier Doppel-Preisträgern in der Geschichte macht.
Sangers Methode basierte auf einem Mix an Insulin-Bruchstücken, den er mit diversen Säuren und Enzymen erzeugte. Sanger zerschnitt die teilweise überlappenden Polypeptide erneut und trennte die Aminosäuren mittels Papier-Chromatographie auf. Dabei markierte er die N-terminalen Aminosäuren mit dem Farbstoff 1-Fluoro-2,4-dinitrobenzol (FDNB, Sangers Reagenz). Hierdurch konnte er sehen, welche Aminosäure wie oft in den Fragmenten markiert war.
Einige Jahre später verbesserte der schwedische Biochemiker Pehr Edman Sangers Technik, indem er FDNB durch Phenylisothiocyanat (PITC) ersetzte. Der Vorteil: PITC markiert nicht nur die N-terminale Aminosäure, sondern induziert beim richtigen pH-Wert auch deren Abspaltung. Das wiederholte Edman so lange, bis alle Aminosäuren nacheinander abgespalten waren. Der Edman-Abbau wurde schnell die vorherrschende Methode zur Protein-Sequenzierung.
Inzwischen hat die moderne Massenspektrometrie (MS) dem Edman-Abbau den Rang abgelaufen. MS-gestützte Analysen ermöglichen es, statt einzelnen Proteinen gleich das ganze Proteom zu identifizieren und zu quantifizieren. Eine typisches MS-Experiment läuft wie folgt ab: Die Proteine werden zunächst enzymatisch zerkleinert. Die resultierenden Peptide werden anschließend ionisiert, in die Gasphase überführt und in einem elektrischen Feld entsprechend ihrem Verhältnis von Masse (Anzahl und Art der Aminosäuren) zu Ladung (Anzahl der geladenen Aminosäurereste) aufgetrennt. Dieses Verhältnis ist für jedes Peptid anders. So verrät die Art der Auftrennung indirekt die Peptidsequenz. Aus den einzelnen Protein-Fragmenten lässt sich dann zurückrechnen, welche Proteine wie häufig in der Probe vorkamen.
Proteom-Analysen sind aus der modernen Biologie nicht mehr wegzudenken. Doch die MS-gestützte Massenabfertigung stößt an ihre Grenzen, wenn Proteine sehr selten vorkommen. „Heutige MS-Technologien erfassen Proteine über vier bis fünf Zehnerpotenzen hinweg. Die Häufigkeit von Proteinen in menschlichen Proben kann aber um ganze zehn Zehnerpotenzen abweichen. Von manchen gibt es weniger als 1.000 Kopien pro Zelle“, illustriert Anne S. Meyer von der University Rochester, UK, das Problem. Die studierte Biologin erforscht Strategien zur sensitiven Protein-Sequenzierung. Um die seltenen Proteine zu erfassen, müssten Forscher sie anreichern. Da man Proteine aber nicht einfach amplifizieren kann, brauche es deutlich mehr Ausgangsmaterial, was laut Meyer oft nicht vorhanden sei.
Hier soll eine neue Technologie Abhilfe schaffen: Die Protein-Sequenzierung auf der Einzelmolekül-Ebene, auch Single Molecule Protein Sequencing (SMPS) genannt. Meyer: „Effiziente SMPS-Methoden könnten die Proteinforschung revolutionieren, wie die NGS-Technologien es mit der Genetik getan haben.“
Starke Worte, doch die Forscherin ist mit ihrer Meinung beileibe nicht allein. Der Wettstreit um die ideale SMPS-Technologie ist längst entbrannt und hat ein ganzes Füllhorn an Methoden hervorgebracht. Mit manchen sequenziert man nur kurze Peptide, mit anderen Proteine oder ganze Proteinkomplexe. Aber nicht nur die Ziele unterscheiden sich, sondern auch die Herangehensweisen.
Protein-Fingerabdruck
Zur Gruppe der Fluoreszenz-basierten Methoden gehört der sogenannte ClpXP-FRET-Ansatz, den Meyers Team mitentwickelt hat (PNAS 115(13): 3338-43). Im Zentrum steht die bakterielle ClpXP-Protease. Zunächst markieren die Wissenschaftler die Proteasen-Oberfläche mit einem Fluorophor und geben dann die Zielproteine hinzu. ClpXP entfaltet und verdaut die Proteine, die ebenfalls an bestimmten Aminosäuren mit Fluorophoren markiert sind. Sobald sich beim Verdau die Fluorophore von Protease und Protein nahekommen, findet ein sogenannter Förster-Resonanz-Energietransfer (FRET) statt. Die FRET-Signale verraten die Reihenfolge der markierten Aminosäuren. „Die ClpXP-FRET-Methode ist ausreichend sensitiv, um auch die Proteine zu detektieren, die bei MS-Analysen durchs Raster fallen. Da ClpXP die Proteine sehr schnell abbaut, bleichen wir die FRET-Fluorophore auch nicht aus“, beschreibt Meyer die Vorzüge. Es gibt jedoch einen Nachteil: Jedes Zielprotein benötigt eine bakterielle Erkennungssequenz, um zur Protease zu gelangen.
Im Reigen der Fluoreszenz-Ansätze erlebt der Edman-Abbau unter dem Schlagwort Fluorosequencing eine ungeahnte Renaissance. Das Grundprinzip ist gleich geblieben, doch statt einem werden bis zu Millionen von Peptiden parallel mittels PITC-Zugabe abgebaut – jeweils immobilisiert und mit Fluorophoren markiert. Die Analyse übernimmt ein Totalreflexions-Fluoreszenz(TIRF)-Mikroskop statt der herkömmlichen Papier-Chromatographie.
Die meisten Fluoreszenz-basierten Methoden haben allerdings ein gemeinsames Manko: Sie liefern ein unvollständiges Bild des Proteins, da nur wenige Aminosäuren markiert werden. Das Ergebnis sind viele kleine Proteinschnipsel, die erst mithilfe von Protein-Datenbanken einen Sinn ergeben. Wer ein Protein von Grund auf, also de novo sequenzieren möchte, braucht andere Lösungen. Schon seit einiger Zeit nutzen Wissenschaftler Nanoporen, um DNA zu sequenzieren. Dank technischer Verbesserungen haben die Nanoporen aber erst vor wenigen Jahren die DNA-Sequenzierung ordentlich durcheinandergewirbelt. Anders als die üblichen Short-Read-NGS-Technologien lesen moderne Nanoporen-Sequenzierer DNA-Stücke von bis zu einer Million Basen. Nicht die einzige Stärke einer Technik, die in den Augen mancher Experten die Post-NGS-Ära einläuten könnte. Von Third Generation Sequencing ist die Rede.
Nur Löcher in der Membran
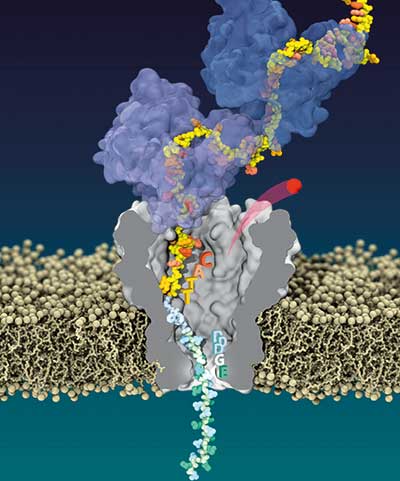
Inzwischen haben auch etliche Proteinforscher die Nanoporen für sich entdeckt. Doch was sind Nanoporen eigentlich und warum sind sie so nützlich? Der Biophysiker Henry Brinkerhoff von der University of Washington, USA, erforscht, wie man Nanoporen für die Protein-Sequenzierung nutzen kann. Er erklärt: „Nanoporen sind nichts anderes als Löcher mit ein paar Nanometern Durchmesser in einer Membran. Für die Sequenzierung platziert man diese Membran zwischen zwei Flüssigkeiten mit einem elektrischen Potenzial. Es entsteht Spannung, die zu einem Stromfluss über die Nanoporen, den einzigen leitfähigen Elementen der Membran, führt.“
Die Sequenzierung läuft dann folgendermaßen ab: DNA-Strang oder Protein werden zuerst linearisiert und durch die Nanopore geführt. Die geringe Größe der Pore verhindert, dass gefaltete Moleküle eindringen können. Beim Durchtritt blockiert jede einzelne Base oder Aminosäure die Pore für einen kurzen Moment und unterbricht den Stromfluss messbar. Dieser elektrische „Abdruck“ ist für jedes Molekül einzigartig. Sobald man alle Signaturen kennt, kann man die Sequenz einfach aus der Stromspur ablesen.
„Nanoporen sind bahnbrechend einfach“, betont Brinerhoff die Stärken der Technik. „Man braucht keine chemischen Reaktionen, man muss die Moleküle nicht markieren oder immobilisieren. Wir isolieren, stabilisieren und messen das Protein in einem Schritt, alles passiert in der Pore.“ Doch während die Nanoporen-Technik bei DNA längst kommerziell erfolgreich und etabliert ist, hinken die Proteinforscher hinterher. Woran liegt das?
Schon das Entfalten der Peptidketten ist eine Herausforderung. Was bei der DNA eine Helikase erledigt, sollen bei Proteinen Chemikalien richten. Doch Harnstoff und Co. denaturieren häufig nicht nur die Zielproteine, sondern auch die Nanoporen selbst, die oft aus porenbildenden Proteinen bestehen. Abhilfe schaffen synthetische Membranen, die keine Proteine enthalten und unempfindlich gegenüber Chemikalien sind. Doch der Nanoporen-Experte schränkt ein: „Synthetische Membranen werden mit winzigen Partikeln beschossen und so durchlöchert. Der Durchmesser der Poren ist aber für jedes Loch ein bisschen anders. Biologische Poren sind dagegen bis aufs letzte Atom identisch, was die Signale viel reproduzierbarer macht. Für mich sind biologische Membranen daher das Mittel der Wahl.“
Doch wie linearisiert man die Proteine zuverlässig, ohne die Biomembranen zu zerstören? Eine Lösung lieferte das Team des US-amerikanischen Biotechnologen Mark Akeson (Nat. Biotechnol. 31(3): 247-50). Es koppelt das Porenprotein α-Hämolysin mit der Unfoldase ClpX. Letztere ist eine Domäne des weiter oben erwähnten ClpXP-Proteasoms. Sie enfaltet die ankommenden Proteine und schleust sie ins Innere des Proteasoms. Die Forscher platzieren die Unfoldase auf der trans-Seite der Nanopore, während die Proteine auf der cis-Seite warten – markiert mit der bakteriellen Erkennungssequenz und einer negativ geladenen Peptidkette. Durch die negative Ladung bewegen sich die Proteine im elektrischen Feld selbständig zur Pore hin, wo sie von der Unfoldase ClpX erkannt und gebunden werden. ClpX entfaltet das Protein und zieht es anschließend durch die Nanopore auf die trans-Seite.
Zwei Funktionen
Die doppelte Funktion der Unfoldase ist ihre große Stärke. Denn Proteine sind, anders als DNA, je nach Sequenz ganz verschieden geladen und bewegen sich im elektrischen Feld nicht automatisch zur Pore hin. Doch Brinkerhoff sieht auch einen Schwachpunkt: „Die Unfoldase zieht das Protein leider nicht gleichmäßig durch die Pore, sondern mit unvorhersehbaren Sprüngen. Das erschwert die Interpretation des gemessenen Signals.“
Der Biophysiker hält eine Strategie für erfolgversprechender, die er als Postdoc im Labor des Nanoporen-Spezialisten Cees Dekkers an der Technischen Universität Delft, Niederlande, mitentwickelte (Science 374 (6574): 1509-13). Brinkerhoff und seine Kollegen nahmen sich die DNA-Sequenzierung zum Vorbild. Das Herzstück ihres Aufbaus ist die DNA-Helikase Hel308. Sie ist an ein kurzes DNA-Stück, den Extender, gekoppelt, der wiederum mit einem DNA-Peptid-Konjugat verbunden ist, das einige DNA-Basen und das Zielpeptid enthält. Im Ausgangszustand wird Hel308 von einem weiteren DNA-Strang blockiert, der in der Membran verankert ist. Sobald die negative DNA-Ladung das DNA-Peptid-Konjugat durch die Nanopore drückt, wird Hel308 aktiviert und beginnt damit, den DNA-Teil des Konjugats zu entwinden. Dabei zieht Hel308 den ganzen DNA-Peptid-Strang gleichmäßig wieder „zurück“ auf die cis-Seite der Pore. Hierbei findet die eigentliche Sequenzierung statt.
Das Prozedere klingt einigermaßen kompliziert. Brinkerhoff beschreibt die Idee dahinter: „Wir haben uns gefragt: Warum nutzen wir nicht einfach ein Enzym, das man seit Langem erfolgreich für die Sequenzierung von DNA nutzt? Hel308 prozessiert den Strang so gleichmäßig, dass wir die Aminosäuren im Signal viel besser erkennen. Da DNA-Helikasen nicht an Proteine binden, benötigen wir die DNA als Ankerpunkt.“
Mit Helikase-Team genauer
Nicht nur die akkurate Helikase sorgt dafür, dass Brinkerhoffs Sequenzierungen zuverlässig sind. Dem Team gelang es, mehrere Helikasen hintereinander an den DNA-Extender zu binden. Die Folge: Sobald ein Sequenzier-Durchgang beendet ist, dissoziiert das erste Hel308-Enzym vom DNA-Strang. Nun wird der Strang wieder in die Pore gezogen und die zweite Helikase am DNA-Extender wird aktiviert, sodass das Ganze von vorne beginnen kann. Auf diese Weise konnten die Forscher Peptide dutzende Male ablesen und die Genauigkeit immer weiter verbessern. „Die Ergebnisse machen mich sehr froh“, ist der Biophysiker erkennbar stolz. „Das könnte ein Meilenstein in der SMPS-Technologie sein. Denn das Hauptproblem bei allen Nanoporen-Techniken ist, die Aminosäuren im elektrischen Signal zweifelsfrei zu identifizieren.“
Das sieht auch die SMPS-Expertin Meyer so: „Wenn es nur vier mögliche Moleküle gibt, A, C, G und T, dann werden Sie die im Signal relativ leicht auseinanderhalten können. Selbst wenn das System nicht perfekt kalibriert ist. Aber zwanzig Aminosäuren sind eine andere Hausnummer. Und diese Moleküle sind hoch divers, teils positiv oder negativ geladen, teils hydrophob und teils verzweigt. Hinzu kommen Phosphorylierungen und andere Modifikationen.“ Um in diesem Chaos den Überblick zu behalten, brauche es akkurate und reproduzierbare Bedingungen. Ob Brinkerhoffs Ansatz wirklich der ersehnte Durchbruch ist, bleibt abzuwarten. Bislang waren die Experimente allesamt auf kurze Peptide beschränkt. Der Weg mag bereitet sein, aber er ist noch lang.
Trotz vieler Hürden ist aber wohl eines sicher: Das Einzelmolekül-Zeitalter bricht auch in der Proteinforschung an. Doch was wird dann aus den altbewährten MS-Analysen? Gilt bald nur noch Klasse statt Masse? Wie wäre es stattdessen mit Klasse und Masse? Sprich: Einzelmolekül-Auflösung und Proteomik kombiniert.
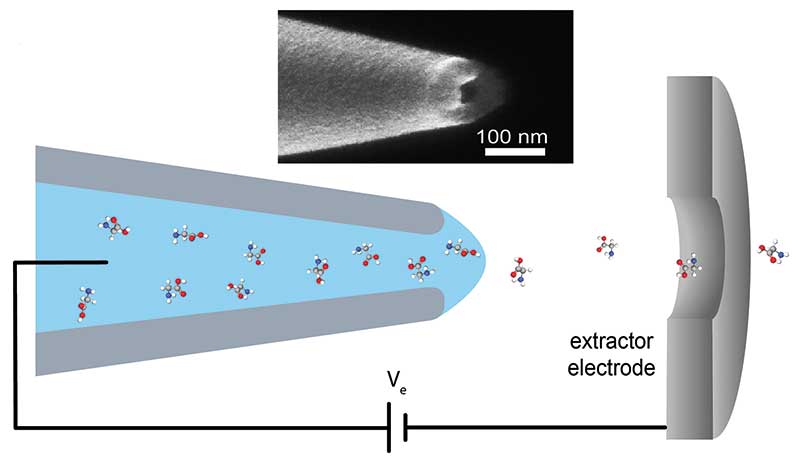
Eine Vision, die Derek Stein von der Brown University in Providence, USA, in die Realität umsetzen möchte. Der Biophysiker ist fest überzeugt vom Potenzial der Massenspektrometrie. Er geht davon aus, dass sich diese auch gegenüber SMPS-Ansätzen behaupten kann und erklärt, wo es aktuell noch hakt: „Sehr viele MS-Systeme setzen die Peptide mittels Elektrospray-Ionisierung (ESI) frei. Das heißt, kleine Tropfen mit Peptid-Ionen werden aus einer Öffnung gesprüht, die Flüssigkeit verdunstet in einem Hintergrundgas und die Ionen gelangen in das Vakuum des Analysators.“ Doch die Tropfen verdunsten nicht schlagartig, sondern in mehreren Zyklen, abhängig von der Größe der Tropfen. Bei jedem Zyklus kommt es ladungsbedingt zu einem physikalischen Phänomen, das als Coulomb-Explosion bezeichnet wird. Dabei gilt: Je mehr Zyklen, umso mehr Coulomb-Explosionen.
Das klingt nach physikalischem Klein-Klein. Doch jede Explosion produziert Ionen-Querschläger, die nicht im Analysator ankommen. Das beeinträchtigt die Ionen-Ausbeute so stark, dass Stein betont: „Wenn wir es schaffen, die Tropfen deutlich kleiner zu halten als bisher, können wir mit jetzigen MS-Techniken tatsächlich eine Auflösung auf Einzelmolekül-Ebene erreichen.“
Steins Team entwickelte eine spezielle Quarzkapillare, die einen Öffnungsdurchmesser von maximal 100 Nanometern aufweist. Die ersten Testläufe mit kleinen Molekülen wie DNA-Basen oder Salzen waren ein voller Erfolg. Die gläserne Nanopore produzierte viel kleinere Tropfen als typische ESI-Öffnungen, wodurch mehr Ionen den Detektor erreichten. Durch den winzigen Durchmesser der Kapillare sank zudem die Flussrate, was sich ebenfalls positiv auf die Ionen-Ausbeute auswirkt. Stein nennt die weiteren Vorzüge seines Systems: „Unser Nanoporen-Elektrospray vereinfacht das Design der MS-Instrumente, mehrere Vakuumstufen sind nicht mehr nötig. Wir benötigen auch kein Hintergrundgas mehr für die Verdunstung, wodurch nochmals mehr Ionen den Analysator erreichen.“
Die MS-Spezialisten arbeiten mit Hochdruck daran, ihr System weiter zu verbessern, um möglichst bald Polypeptide mit der neuartigen Nanopore zu ionisieren. Aktuell steckt ein Manuskript von Steins Gruppe, in dem sie einzelne Aminosäuren testet, im Peer Review-Prozess. Klingt spannend. Und was sagen die Daten? Der Co-Autor hält sich lieber bedeckt, er möchte den Fachgutachtern nicht vorgreifen. Da müsse man leider bis zur Veröffentlichung warten.
Schade, aber verständlich. In der SMPS-Forschung tut sich momentan so einiges – da möchte man lieber nicht zu viel über seine Daten verraten.