Proteomik enthüllt: Proteine organisieren sich zu Quinärstrukturen
Henrik Müller
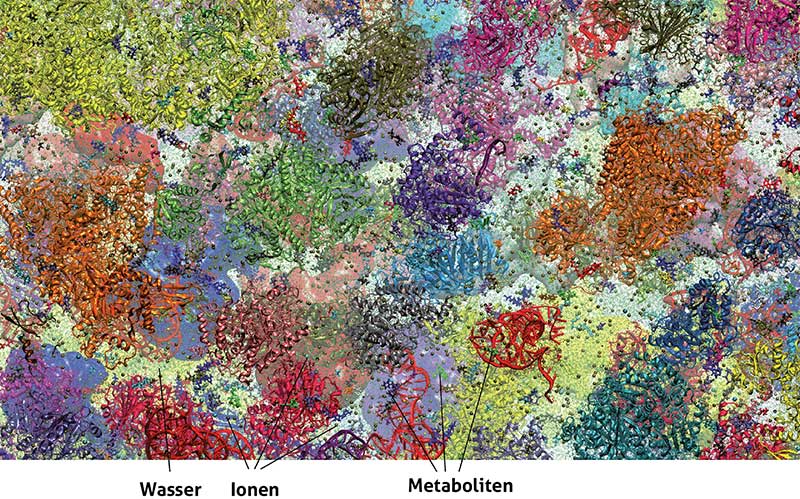
Im Tohuwabohu des Zellinneren interagiert alles mit allem. Flüchtige Wechselwirkungen bereiten mehr als nur den Hintergrund biochemischer Reaktionen, werden für In-Vitro-Analysen aber meist „herausgereinigt“. Vernachlässigen wir jedoch diese transienten Interaktionen, werden wir das Wechselspiel zeitlicher und räumlicher Komplexität in Zellen nicht verstehen.
Ein Protein wird geboren und schiebt sich aus seinem ribosomalen Uterus Aminosäurerest für Aminosäurerest hervor. Nach Anfinsens Dogma besitzt es nur eine native Konformation minimaler freier Energie, die durch seine Aminosäuresequenz vorgegeben ist. Es plumpst also sorglos den Faltungstrichter hinunter, formt seine Extremitäten und gewinnt Gestalt. Mit etwas Glück leisten ihm eineiige Geschwisterchen gleich Beistand. Kurz nach seiner Geburt hat es seine unabänderliche Form erlangt, Sekundär- bis Quartärstruktur sind ausgeprägt. Es ist bereit für seine Aufgabe...
Weit gefehlt! Aus dem Nichts prasseln die enthalpischen und entropischen Komponenten seiner überbevölkerten Umgebung auf den Neuling ein. Und verleihen ihm durch Wasserstoffbrücken, Van-der-Waals-Kräfte, hydrophobe und elektrostatische Wechselwirkungen wieder neuen Ausdruck. Denn die zelluläre Umgebung ist vollgestopft, hochviskos und inhomogen. Räumlich und zeitlich komplex. Der Großteil der Kontakte sind nur flüchtige Begegnungen marginaler Affinität, die in ihrer Häufigkeit spezifische Bindungsereignisse aber weit übertreffen. Den kombinierten Einfluss dieser Effekte fasste Edwin McConkey bereits 1982 unter dem Begriff Quinärstruktur zusammen (PNAS 79: 3236-40).
Schnell lernt jedes frischgeborene Protein daher drei harte Lektionen:
- Es darf nicht überall hin,
- nirgendwo kommt es schnell hin, und
- sein Weg ist steinig.
Zelluläre Diskriminierung mag ungerecht sein. Mit 200 bis 400 mg/ml an Makromolekülen in einer Zelle beschränkt deren Crowding aber, wo ein Protein bestimmter molekularer Masse hin kann. Ist es zu groß, zu unflexibel oder nicht formschön genug, sich unter seinesgleichen zwischen den Cytoskelett-Filamenten durchzuquetschen, hilft alles Wettern nichts. Es mag nur einen Teil des noch vorhandenen Platzes jemals besuchen können. Wer sich für die Theorie dahinter interessiert, dem sei das Asakura-Oosawa-Modell (J. Cell Biol. 175: 681-6), die Flory-Huggins-Theorie (PNAS 111: 4874-9) und die Theorie der skalierten Teilchen (Biopolymers 10: 2093-120) ans Herz gelegt.
Auf zellulärer Ebene resultiert Crowding in einem Volumenausschlusseffekt. Dessen Entropiekomponente treibt hochmolekulare Komplexe wie etwa Substrat-kanalisierende Metabolons zusammen und führt zu membranloser Kompartimentierung – bestimmt also letztlich die biochemische Organisation ganzer Zellen.
Volumenausschluss nimmt Proteinen aber nicht nur Möglichkeiten. Ein strukturschwaches Protein leidet unter erhöhtem Trägheitsradius. Und damit auch unter erhöhtem beanspruchten Volumen. Bei Enge hat das Protein aber keinen Platz zur Entfaltung und bleibt stabil.
Crowding kann den Unterschied machen
Der Biochemiker Volker Dötsch von der Goethe-Universität Frankfurt am Main erklärt: „Für ungefaltete Sequenzen ist eine helikale Konformation kompakter als die entfaltete Struktur. Allerdings muss die Sequenz dazu eine intrinsische Neigung zur Helix-Bildung haben. Sequenzen, die gelöst in einem Gleichgewicht aus teilweise helikaler Struktur und ungefaltetem Zustand vorliegen, werden durch Volumenausschluss in eine kompaktere Struktur überführt.“ Crowding verringert also die Stabilität ungeordneter Strukturen. Besagt zumindest die Theorie.
Praktisch kam dem Simon Ebbinghaus im Jahr 2011 auf die Schliche, damals noch Postdoktorand bei Martin Gruebele an der University of Illinois in Urbana-Champaign/USA, als er mit seinen Kollegen die Stabilität von Phosphoglyceratkinase (PGK) mittels Fast Relaxation Imaging (FReI) untersuchte. FReI beruht auf dem optischen Nanometermaß des Förster-Resonanzenergietransfers (FRET), kombiniert Fluoreszenzmikroskopie einzelner Zellen aber mit einem plötzlichen Temperatursprung. Dafür verschiebt ein Infrarotlaser das Faltungsgleichgewicht FRET-markierter Proteine und eine CCD-Kamera nimmt die FRET-Effizienz während Denaturierung und Rückfaltung auf. Ebbinghaus fand, dass entartete Knochengewebszellen, Adenokarzinomzellen von Henrietta Lacks (HeLa-Zellen) wie auch E. coli-Zellen die PGK um bis zu 2 kcal/mol stabilisieren (J. Phys. Chem. Lett. 2: 314-19).
Das ist eine Menge. Der Unterschied in der freien Energie eines Proteins zwischen nativem und denaturiertem Zustand liegt nur bei 5 bis15 kcal/mol. Crowding kann also den Unterschied machen. Später stellte sich allerdings heraus, dass neben PGK etwa auch die B1-Domäne von Protein G (GB1) durch zelluläre Umgebungen stabilisiert wird, nicht aber das Oberflächenprotein VlsE, Chymotrypsin-Inhibitor 2 oder die Superoxid-Dismutase 1 (SOD1). Ebbinghaus, mittlerweile Professor für Physikalische und Theoretische Chemie an der TU Braunschweig, fand, dass SOD1 durch quinäre Wechselwirkungen sogar destabilisiert wird (J. Am. Chem. Soc. 141: 4660-69).
Es ist also nicht ganz so einfach, wie durch Volumenausschluss vorhergesagt. Selbst energetisch ähnliche Proteine können in vivo verschieden sein. Ebbinghaus‘ Doktorandin Sara da Silva Ribeiro erklärt dazu: „Dafür haben wir noch keine endgültige Antwort. Die hohen intrazellulären Konzentrationen an Makromolekülen resultieren in zwei gegenläufigen Prozessen: einem entropischen Volumenausschlusseffekt sowie enthalpischen quinären Interaktionen. Volumenausschluss stabilisiert Proteine. Quinäre Wechselwirkungen tragen zur Stabilisierung bei, falls sie eine abstoßende Wirkung entfalten. Haben sie eine anziehende Wirkung, können sie den Volumenausschluss ausgleichen und destabilisierend wirken. Das bedeutet, dass jedes Protein einzeln zu betrachten ist. Alles hängt von der chemischen Natur interagierender Reste auf der Proteinoberfläche ab.“
Das überrascht ein juveniles Protein nicht. Denn egal, ob die zelluläre Suppe es stabilisiert oder nicht, seine zweite Lektion ist, dass es nirgendwo schnell hingelangt. Es klebt an anderen Molekülen, und zwar aufgrund von Elektrostatik seiner Oberflächenladungen und Hydrophobizität, also seinem Einfluss auf die Unordnung von Solvathüllen. All das steigert die Viskosität einer Lösung exponentiell.
Selbst flinke Wassermoleküle werden langsamer
Wie Strukturen höherer Ordnung zu einem hochviskosen Zellinneren führen, überprüften der Biophysiker Gary Pielak und seine Mitarbeiter am Department of Chemistry der University of North Carolina in einer Reihe von NMR-Studien. Zum 7,4 kDa Chymotrypsin-Inhibitor 2 (CI2) gaben sie jeweils 300 mg/ml des synthetischen Polymers PVP, des Sucrose-Polymers Ficoll, von Lysozym (15 kDa), Ovalbumin (43 kDa) oder BSA (67 kDa). Sowohl die Polymere als auch die Proteine reduzierten CI2s translationale Diffusion um mehr als einen Faktor Zehn, seine Rotationsdiffusion um das Fünf- bis Zehnfache (J. Am. Chem. Soc. 132: 9392-7).
Interessanterweise beeinflussten die künstlichen Polymere die translationale Diffusion mehr als die rotationale, während die proteinogenen Crowder den entgegengesetzten Effekt hatten – und zwar unabhängig von ihrer Größe und Ladung. Das erlaubt zwei gewichtige Schlussfolgerungen. Erstens taugen synthetische Polymere wenig, um die Ungleichmäßigkeit des intrazellulären Milieus zu simulieren. Zweitens sind es schwache, unspezifische, nicht-kovalente, kurzum quinäre Wechselwirkungen zwischen CI2 und den Crowding-Proteinen, die seine zelluläre Diffusion hemmen. Selbst flinke Wassermoleküle werden durch Protein-Crowding um 25 Prozent verlangsamt (PLoS Comput. Biol. 12: e1005040).
Erneut steckt hier der Teufel im biomolekularen Detail. Im Vergleich zum globulären CI2 verhält sich das intrinsisch ungeordnete, 14 kDa große α‑Synuclein gegensätzlich. In verdünnter Lösung diffundiert α‑Synuclein halb so schnell wie CI2. In Gegenwart der obigen Crowder schaltet es aber trotz erheblich größeren hydrodynamischen Volumens in den nächsten Gang. Es translatiert bis zu doppelt so schnell wie CI2. Pielak und Co. schlussfolgerten daher, dass auch die Proteinform auf das Diffusionsverhalten einwirkt und demnach bei der Analyse quinärer Interaktion berücksichtigt werden muss (J. Phys. Chem. Lett. 3: 2703‑6).
Und die Rotationsdiffusion? Bevor Lila Mary Gierasch 2016 Chefredakteurin des Journal of Biological Chemistry wurde, untersuchte sie globuläre Proteine mittels In‑Cell-NMR-Spektroskopie. Ihre Testobjekte GB1, NmerA und Ubiquitin ähneln sich in Molekulargewicht (6‑9 kDa) und 3D-Struktur (ein bis zwei α‑Helices gegenüber einem β‑Faltblatt), unterscheiden sich in E. coli aber maßgeblich in ihrem Rotationsverhalten. Gierasch und ihr Team an der University of Massachusetts folgerten, dass dafür ein kniffliges Zusammenspiel variabel verteilter Oberflächenreste von unterschiedlicher Elektrostatik und Hydrophobizität verantwortlich zeichnet. Oder kurz gesagt, dass die individuelle Proteinsequenz entscheidet, wie klebrig sie eine Umgebung befindet. Ganz zu schweigen davon, dass Proteine auf unterschiedliche Quinärstrukturen in unterschiedlichen Kompartimenten unterschiedlich reagieren können. Ein wunderschönes Beispiel hierfür präsentierte Martin Gruebele, Ebbinghaus‘ Postdoc-Betreuer, vor drei Jahren in den FEBS Letters ( 590: 1409-16).
Ständiges Bombardement und Kollisionen
Derartige Einzigartigkeit hilft einem juvenilen Protein aber nicht weit, lernt es in seiner dritten Lektion neben dem Zuckerbrot doch auch die Peitsche kennen. Denn trotz Klebrigkeit der cytoplasmatischen Suppe ist es einem ständigen Bombardement von Wassermolekülen ausgesetzt. Bei physiologischer Temperatur übertragen Wasserkollisionen Energiefluktuationen von 0,01 kcal/mol auf ein Protein. Das klingt wenig. In einer überfüllten Umgebung sind Molekülbewegungen aber oft korreliert. Zum Beispiel kollidieren verdrängte Wassermoleküle gleichzeitig mit Interaktionspartnern und verlangsamen dadurch Bindungskinetiken. Simulationen der Moleküldynamik haben gezeigt, dass hydrodynamische Effekte die Assoziationsrate von Proteinen um 35 bis 80 Prozent herabsetzen (Biophys. J. 99: L75‑7). Zugleich stabilisieren sie übrigens aber auch die Schnittstelle zweier Proteine um bis zu 1 kcal/mol.
Diese Energiebeträge liegen erneut in der gleichen Größenordnung wie die thermodynamische Proteinstabilität selbst. Könnten quinäre Wechselwirkungen folglich die Tertiärstruktur eines Proteins ändern? Volker Dötsch meint: „Bisher gibt es keine handfesten Hinweise darauf. Die 3D-Struktur einer gefalteten Domäne werden sie nicht ändern.“
Im Jahr 1973 publizierte der Nobelpreisträger Christian B. Anfinsen seine thermodynamische Hypothese der Proteinfaltung (Science 181: 223-30). Seitdem ist sie unwiderlegt. Auch wenn Prionen, metamorphe Proteine und Chaperone das Dogma herausforderten. Dötsch schließt: „Proteine tragen in ihrer Sequenz alle notwendige Information, so dass sie ihre 3D‑Struktur annehmen können – zumindest im Prinzip. Denn viele globuläre Proteine brauchen dafür Chaperone. Wir müssen Anfinsens Dogma daher nur leicht modifizieren, quasi Fußnoten anfügen, um zu sagen, dass es Co-Faktoren braucht, die bei der Faltung helfen.“
Ein anderes Kaliber als Faltungshelfer stellen intrinsisch ungeordnete Proteine (IDP) dar. Denn unstrukturierte Bereiche sind anfälliger für quinäre Einflüsse als biologisch inerte Proteindomänen. Dötsch erörtert: „Die Faltung von IDPs hängt stark von ihrer Umgebung ab. Ein klassisches Beispiel dafür ist die ungefaltete Transaktivierungsdomäne des Tumorsuppressors p53. Erst Bindung an ihren Inhibitor Mdm2 oder an Co-Faktoren wie die Acetyltransferase p300 induziert eine α‑helikale Struktur. Auch β‑Faltblätter sind möglich. Beispielsweise erweitern die LIR-Sequenzen in Autophagie-Rezeptoren die bestehenden β‑Faltblätter von Autophagie-Modulatoren wie LC3 und GABARAP um einen Strang. Interaktionspartner induzieren also funktionale Sekundärstrukturelemente, die dann zu einer affineren Bindung führen.“
Proteinstrukturen in der Zelle analysieren
Demzufolge können nur spezifische Interaktionen die IDP-Struktur ändern? Dötsch dazu: „Nein, es gibt eine Publikation aus der Anfangszeit der In‑Cell-NMR aus Pielaks Labor. Die Kollegen zeigten, dass auch molekulares Crowding eine höhere Kompaktheit in einem Protein induzieren kann.“
Das bezeichnete Protein ist der negative Regulator der Flagellin-Synthese (FlgM) aus Salmonella typhimurium. Tatsächlich ist es das einzige bisher beobachtete IDP, das durch zelluläres Crowding Struktur gewinnt. Das NMR-Spektrum einer verdünnten FlgM-Lösung zeigt die enge Dispersion chemischer Verschiebungen und scharfen Resonanzlinien eines unstrukturierten Proteins. In E. coli passiert aber Erstaunliches. Während sich in der N-terminalen Hälfte gar nichts tut, bildet die C‑terminale Hälfte zwei α-Helices aus. Und zwar nicht nur, wenn es seinen natürlichen Interaktionspartner, den Transkriptionsfaktor σ28, bindet, sondern auch ohne σ28 in Gegenwart von 400 mg/ml der Crowding-Reagentien Glucose, BSA oder Ovalbumin. Offensichtlich diktieren neben spezifischen Bindungen auch quinäre Interaktionen die Struktur von IDPs.
Wenn also die zelluläre Umgebung strukturell relevant ist, steckt dann doch nicht alle notwendige Information in der Primärsequenz eines IDPs? Dötsch resümiert: „Nach Anfinsen müsste man den Sequenzraum einfach nur erweitern. Die 3D-Struktur ist nicht mehr nur im Peptid kodiert, sondern in Peptid plus Interaktionspartner. Dann passt das Dogma wieder.“ Aber das Crowding-Reagenz Glucose ist doch kein Protein? „Glucose induziert nur eine Helix, bildet aber keine globuläre 3D-Struktur aus. Außerdem wurde die Entstehung einer helikalen FlgM-Struktur nicht auf der Basis von NMR-Daten, sondern von CD-Spektroskopie gefolgert. Ob sich die α‑Helix von der durch σ28 induzierten unterscheidet, lässt sich nicht sagen.“ Anfinsens Dogma scheint weiterhin sicher.
Derartige Untersuchungen sind keine leichte Aufgabe. Thermodynamische und kinetische Parameter in lebenden Zellen sind schwierig experimentell zu fassen. Wie es klappen kann, erörtern Dötsch und Mitarbeiter in einem Review (Angew. Chem. Int. Ed. 53:10300-14). „Vor zwanzig Jahren haben wir in San Francisco die ersten NMR-Experimente in lebenden Zellen gemacht, damals noch in E. coli. Die Wechselwirkungen in der Zelle waren viel umfangreicher als in vitro. Wegen der Dichte an Makromolekülen natürlich keine Überraschung. Doch um Wechselwirkungen zu identifizieren, die in vitro nicht da sind, ist In-Cell-NMR bestens geeignet.“
Nicht, dass es damit keine technische Herausforderung mehr wäre. Dötsch erklärt: „Zugrunde liegt ein intrinsisches Problem der Flüssig-NMR, das uns schon in vitro Grenzen setzt. Alles hängt davon ab, ob ein Protein Signale zeigt. Je höher seine Rotationskorrelationszeit, also je langsamer es sich aufgrund seiner Größe bewegt, desto breiter werden seine Signale. Bis sie irgendwann verschwinden. Wenn In-Vivo-Interaktionen, egal ob spezifisch oder nicht, das apparente Molekülgewicht erhöhen, geht auch die Rotationskorrelationszeit weiter hoch. Leider sind deshalb die meisten Proteine in vivo nicht detektierbar.“
Die Technik setzt weiterhin Grenzen
Weshalb es noch weitere zehn Jahre dauerte, bis die erste und bis vor kurzem einzige Struktur eines Proteins in vivo aufgeklärt werden konnte (Nature 458: 102-5). Schlüssel dazu war eine außergewöhnlich hohe Expressionsrate des Metall-bindenden Proteins TTHA1718 von 4 mM. Erst seit zwei, drei Jahren reichen Proteinkonzentrationen im zweistelligen µM‑Bereich zur Strukturaufklärung in Xenopus-laevis-Oocyten aus, wodurch eine weitere Hürde intrazellulärer NMR-Forschung in Angriff genommen wird. „In‑Cell-NMR mit physiologischen Proteinkonzentrationen werden wir in absehbarer Zukunft nicht machen können“, gibt Dötsch zu, der vor allem als Experte zellfreier Expressionssysteme bekannt ist. Er ergänzt: „In-Cell-NMR ist noch keine Standardtechnik. Relativ leicht können wir aber den physikalischen Zustand eines Proteins in vivo qualitativ analysieren, ebenso wie Crowding-Effekte, Bindungskinetiken und vor allem enzymatische Kopplungen. Beispielsweise lassen sich die Kinetiken posttranslationaler Modifikationen wunderbar messen. Mittlerweile sehen wir, wo und wann innerhalb des Zellzyklus Phosphoryl- oder Acetylgruppen angebracht werden. Und über solche NMR-basierten Kinetikstudien können wir sehr viel über sehr interessante Biologie lernen.“
Neben Xenopus-laevis-Oocyten, deren Größe die Mikroinjektion von Proben erlaubt, werden inzwischen auch Säugerzellen verwendet, in die Biomakromoleküle durch Poren-bildende Agenzien wie Streptolysin O, Zell-penetrierende Peptide oder Transfektion von DNA-Plasmiden eingebracht werden.
Trotz NMR-Begeisterung vergisst Dötsch nicht, die Schwachstellen der Technik zu erwähnen: „In-Cell-NMR ist bestens für die Untersuchung von Konformationen geeignet, kann aber über deren Lokalisation keine Aussage treffen. Komplementär dazu ist etwa Fluoreszenzspektroskopie, mit der wir die Bewegung eines Proteins beobachten können.“
Zusätzlich zu Flüssig-NMR und Fluoreszenzspektroskopie gibt es weitere Techniken, die bei der Aufklärung intrazellulärer Details mitmischen. Festkörper-NMR schockgefrorener Zellen etwa leidet nicht unter den Größenbegrenzungen der Flüssig-NMR, aber dafür unter noch ärgeren Sensitivitätsproblemen. Mit der hochsensitiven Elektronenspinresonanz (EPR)-Spektroskopie können intramolekulare Distanzen, beispielsweise bei Konformationsänderungen, begutachtet werden – allerdings nur bei kryogenen Temperaturen von 50 Kelvin beziehungsweise -223 Grad Celsius. Der letzte Schrei sind Bioreaktoren, die Zellen während der NMR-Messung mit frischem Medium versorgen und konstante Level an O2 und CO2 aufrechterhalten, wenn bisher auch nur für fünf bis sechs Stunden.
Der Trend ist trotzdem klar. Die Forschungsgemeinde ist sich einig, dass quinäre Interaktionen die thermodynamische Stabilität des Proteoms bestimmen sowie Komplexität und Dynamik des Zellinneren organisieren. In ihrem letzten Review gehen Ebbinghaus et al. sogar einen Schritt weiter (FEBS Lett. 592: 3040-53). Mit den Worten der Erstautorin Sara da Silva Ribeiro: „Quinäre Interaktionen sind keine unspezifischen Wechselwirkungen. Da sie Zellen eine Funktion bereitstellen, sind sie molekularer Evolution unterworfen. In diesem Sinne sind sie spezifisch“.
Ruf nach Standardisierung
Wie biologisch relevant das ist, zeigen etwa membranlose Organellen. In deren Inneren halten quinäre Interaktionen Proteine konformationell stabil, aber gleichzeitig auch flexibel genug, um eine Art Flüssigphasen-Mikroreaktor zu erschaffen. Wenn bei deren Reifung etwas schiefgeht, drohen neurodegenerative Erkrankungen wie Morbus Alzheimer. (Ein Beispiel dazu findet sich in dieser LJ-Ausgabe auf Seite 32: Link).
Den Bedürfnissen intrazellulär interessierter Biowissenschaftler hinkt einzig die Technik hinterher. In der Zwischenzeit ist deshalb die Quinary Structure Assessment (QSA) Initiative von Philipp Selenko, Leibniz Institut für Molekulare Pharmakologie (FMP) in Berlin, eine bestechende Idee. Ähnlich der Kooperation innerhalb intrazellulärer Proteinkomplexe ruft Selenko in einem zwei Monate alten Review (Int. J. Mol. Sci. 20: E1278) alle NMR-Gruppen auf, Spektren ihrer Lieblingsproteine unter standardisierten Bedingungen in Puffer versus E. coli- und HeLa-Lysaten aufzunehmen und zu vergleichen. Ein solcher Vergleich wäre ein Meilenstein in der Erforschung quinärer Interaktionen. Denn in einem herrscht inzwischen besondere Klarheit: Im Teströhrchen verdünnte Proben, wie bei der Untersuchung von meist weniger als 1 mg/ml gereinigten Proteins, entsprechen der ursprünglichen zellulären Umgebung rein gar nicht.
Letzte Änderungen: 29.11.2019