Künstliche Intelligenz im Labor
Lernfähige Meister der Daten
Mario Rembold
Künstliche Intelligenz (KI) und Deep Learning bringen nicht nur Google, sondern auch die Lebenswissenschaften voran. Sie sind zwar kein Allheilmittel – doch ohne sie kommt die Forschung immer weniger aus, wenn es um große und komplexe Datensätze geht.
„Jede hinreichend fortschrittliche Technologie ist von Magie nicht zu unterscheiden“, stellte der Physiker und Science-Fiction-Autor Arthur C. Clarke einst fest. Und mal ganz unter uns: Wer hat sich nicht schon mal gefragt, ob das eigene Smartphone wohl Gedanken lesen kann? Dann zum Beispiel, wenn Ihr Display Sie über den nächsten Bus nach Hause benachrichtigt – genau in dem Augenblick, in dem Sie am späten Abend den Bürorechner runtergefahren und den Mantel übergezogen haben, um das Institut zu verlassen.
Deep Learning und Künstliche Intelligenz (KI) – das sind Schlagworte, die wir dazu gerade lesen. Auch in den Lebenswissenschaften spielen schlaue und lernfähige Algorithmen eine immer größere Rolle. Das Grundprinzip: Die Programme erkennen Muster in großen Datensätzen und ziehen eigenständig Schlussfolgerungen. In anderen Fällen zeigt der Entwickler seiner Software Bilder oder Sprachschnipsel, denen bestimmte Wörter oder Eigenschaften zugeordnet sind – und nach einer Trainingsphase kann die App auf einmal gesprochene Sätze verstehen, oder Ihre Smartphone-Kamera wartet mit dem Foto, bis Sie lächeln. Man könnte wirklich an Zauberei glauben.
Etwas nüchterner blickt Thomas Berlage auf den technischen Fortschritt in Sachen KI: „Für uns Mathematiker ist das alles pure Statistik.“ Und in der biomedizinischen Forschung sei daran auch nichts prinzipiell Neues. „Jede Leitlinie, die in der Medizin zur Anwendung kommt, sollte ja eigentlich auch das Ergebnis einer ausgiebigen Datenanalyse sein“, so Berlage. Je größer die Patientenzahlen und Datenmengen einer Studie, desto schwerer wird es aber, von Menschenhand nach Korrelationen und Mustern zu suchen. „Hinter jeder Genomanalyse steckt Bioinformatik, und die Übergänge zu dem, was man heute hauptsächlich mit KI meint, sind fließend.“
Auf das Training kommt‘s an
Berlage koordiniert das Geschäftsfeld Life Sciences & Health Care im Fraunhofer-Projekt „Big Data und Künstliche Intelligenz“. Er forscht am Fraunhofer-Institut für Angewandte Informationstechnik in Sankt Augustin sowie an der Rheinisch-Westfälischen Technischen Hochschule (RWTH) Aachen. Wann immer ein neuer Algorithmus oder eine neue Art der Datenverarbeitung entwickelt wird – für Berlage zählt dabei nicht allein die Kreativität der Programmierer. „Wir müssen auch nachweisen, dass die Anwendung einen Nutzen hat und bessere Ergebnisse liefert als andere Techniken“, betont er. Ein lernender Algorithmus kann nämlich auch seine Tücken haben, gerade wenn er auf den ersten Blick perfekt zu funktionieren scheint.
„Der Computer übernimmt natürlich auch alle Eigenheiten, die der Trainingsdatensatz aufweist“, so Berlage. Nehmen wir als Beispiel, dass man bei Patienten Hautverfärbungen zuverlässig auf einer Skala von „unbedenklich“ bis „wahrscheinlich bösartig“ einteilen möchte. Bekommt das Programm nun einen Trainingsdatensatz, in denen bereits fehlerhafte Zuordnungen (Annotationen) enthalten sind, wird er auch diese Zusammenhänge lernen. Wichtig ist also, dass man die trainierte KI zusätzlich mit Testdatensätzen auf die Probe stellt, die sie noch nicht kennt. Doch selbst mit makellosen Trainingsdaten kann ein Algorithmus später versagen. Bleiben wir beim Beispiel Haut-Screening: Bekommt das System zum Training allein Daten junger mitteleuropäischer Männer mit heller Haut zu sehen, so darf man sich nicht darauf verlassen, dass der Computer bei einer dunkelhäutigen Frau aus Zentralafrika zuverlässige Voraussagen liefert.
„Wer mit Machine Learning anfängt, macht solche Erfahrungen sehr schnell“, berichtet Berlage. „Man trainiert mit einem Datensatz, und bei ähnlichen Datensätzen klappt alles perfekt. Doch hat man später einmal mehr Daten zur Hand, funktioniert es auf einmal weniger gut.“ Bei der Entwicklung eines KI-Systems sieht Berlage daher mehr als 80 Prozent des eigentlichen Aufwands darin, an qualitativ hochwertige und zuverlässige Daten zu kommen.
Speziell für die Onkologie glaubt Berlage, dass KIs künftig eine größere Rolle spielen werden. „Da gibt es lernbasierte Algorithmen, die durchaus in der Liga der Ärzte mitspielen“, ist er sicher – stellt aber auch klar: „Kein Mediziner muss derzeit Angst haben, von Maschinen ersetzt zu werden.“ Ein Computersystem kann den Arzt bei seinen Entscheidungen unterstützen, weil es aus hunderttausenden Patientendaten gelernt hat. Doch den Gesamtkontext muss am Ende der Mensch beurteilen. „Es kann sein, dass diese Systeme irgendwann so gut sind, dass wir im Zweifelsfall die Diagnose des Computers als die bestmögliche Einschätzung akzeptieren und Leitlinien dementsprechend anpassen.“ Selbst dann käme aber immer wieder neues Wissen aus der Forschung nach, worauf KIs angepasst und neu validiert werden müssten. Ohne den menschlichen medizinischen Sachverstand wäre hier folglich kein Fortschritt möglich.
KIs in der Biomedizin sind aber schon jetzt mehr als bloß Proof of Concept. „Zum Beispiel haben wir für Bayer ein System entwickelt, das Krebszellen automatisch klassifiziert“, verrät Berlage. Das Tool soll die Suche nach Wirkstoffen gegen Krebs erleichtern, indem es teilungsaktive Zellen von toten oder sich langsam vermehrenden Zellen unterscheidet.
Auch bei Roche Diagnostics arbeitet man mit Bilderkennungssoftware, wie uns Johannes Ritter, Kommunikationsleiter am Standort Penzberg, mitteilt. „Monoklonale Zelllinien können heute bereits mithilfe von automatischer Bilderkennung und mittels Deep Learning gefiltert werden“, berichtet er im Zusammenhang mit der Entwicklung therapeutischer Antikörper bei Roche. Und für Studien zur Krebs-Immuntherapie annotieren die Forscher Bilder aus Tumor-Biopsien und lassen sich dabei von einer KI unterstützen.
Novartis führt derzeit Daten aktuell laufender sowie in der Vergangenheit abgeschlossener klinischer Studien zusammen, um Abläufe bei der Medikamentenentwicklung zu optimieren. So sollen Kosten für künftige Studien besser vorhergesagt werden, ebenso wie Studiendesign und Probandenrekrutierung. Auch laufende Projekte wollen die Pharma-Entwickler so besser im Blick behalten – Nerve Live nennt Novartis ihre neue datengetriebene Entwicklungsplattform.
KI und Deep Learning sind also schon im Alltag vieler Pharmafirmen angekommen. Bis der Computerassistent in der Hausarztpraxis oder im Krankenhaus zum allgemeinen Standard wird, dürfte es aber noch einige Jahre dauern; schließlich müssen hier Vor- und Nachteile sorgfältig abgewogen und die Techniken zuverlässig standardisiert und validiert sein.
Mikroskopier-Helfer
Tatsächlich sind viele Konzepte hinter KI und Deep Learning schon vor Jahrzehnten entwickelt worden. Allerdings scheiterte die Arbeit ab einem gewissen Punkt einfach an der Rechenleistung – so etwa mit künstlichen neuronalen Netzen. Heute jedoch können Programmierer am Heimrechner ausprobieren, wovon Informatiker in den Siebzigerjahren nur träumen konnten. Mit immer leistungsfähigeren Computern wuchsen seit Beginn des Jahrtausends beispielsweise auch die Datenarchive aus Omics-Projekten. Zudem wurde Speicherplatz als limitierender Faktor immer unbedeutender, sodass Forscher heute massenhaft Bilddaten generieren und aufbewahren können – zum Beispiel beim Mikroskopieren. Allerdings stellt sich auch hier die Frage: Welcher Mensch soll das alles auswerten?
Besuchen wir das BIOSS (Center for Biological Signalling Studies) der Uni Freiburg. Dort leitet Thorsten Falk die Core Facility für Bildanalytik und hat mit seinen Mitarbeitern ein Tool zur Zellsegmentierung namens „U-Net“ entwickelt. Zellsegmentierung, damit ist das Identifizieren von Zellen in mikroskopischen Aufnahmen gemeint. Dem menschlichen Auge helfen zum Beispiel DAPI-Färbungen des Zellkerns, um echte Zellen von anderen Partikeln in der Probe zu unterscheiden. „In vielen Fällen braucht U-Net solche Fluoreszenzmarkierungen gar nicht“, freut sich Falk über die Vorteile der Software. „Natürlich muss man sich einmal die Arbeit gemacht haben, Bildmaterial selber anzuschauen und zu annotieren“, fährt er fort. Denn zunächst braucht U-Net, klar: Trainingsdaten.
„Ursprünglich hat Olaf Ronneberger U-Net programmiert“, blickt Falk zurück, „das war 2015“. Die ersten Versionen musste man über die Kommandozeile aufrufen – ein Weg, den nicht alle User als nutzerfreundlich empfinden. Inzwischen gibt es eine Neuauflage, an der auch Falk mitprogrammiert hat. „Diese Version lässt sich als ImageJ-Plugin verwenden.“ ImageJ ist ein quelloffenes Bildverarbeitungsprogramm mit einer grafischen Oberfläche. „ImageJ bietet viele Funktionen und eben ein starkes Plugin-Interface; daher ist es in der Community sehr beliebt“, weiß Falk. Und über das Plugin steht U-Net nun auch Wissenschaftlern zur Verfügung, die sich nicht mit Kommandozeilen-Befehlen herumschlagen wollen.
Zellzähler
Falk geht auf einen weiteren Vorteil in Sachen Nutzerfreundlichkeit ein: „U-Net ist schon vortrainiert mit Datensätzen verschiedener Zelltypen und unterschiedlichen Aufnahmemodalitäten.“ Für eigene Zellsegmentierungs-Aufgaben kann man das Modell nachtrainieren, doch dafür reichen in der Regel weniger als zehn Bilder – so zumindest schreiben es Falk und Kollegen im Anfang des Jahres bei Nature Methods erschienenen Artikel, in dem sie das U-Net-Plugin für ImageJ vorstellen (16(1): 67-70). Falk räumt aber ein, dass die Standardvariante von U-Net allein für die binäre Zellsegmentierung geeignet ist – also für die Einteilung von Strukturen in die Kategorie „Zelle“ oder „keine Zelle“. Hilfreich ist diese Funktion, wenn man aus großen Bildarchiven Zellen auszählen will. „Aber selbstverständlich kann man mit eigenen Datensätzen weitere Fähigkeiten trainieren“, ergänzt Falk.
Denkt man an das Problem der Reproduzierbarkeit in den Lebenswissenschaften, so werfen lernende Algorithmen neue Fragen auf. Klassischerweise gibt man im „Material-und-Methoden“-Teil ein statistisches Verfahren an oder verweist auf den Quellcode einer Software – und andere Forscher können dann testen, ob sie mit ihren Daten und der gleichen Auswertung ähnliche Ergebnisse bekommen. Doch bei einer Anwendung wie U-Net sagt der Programm-Code wenig aus. Entscheidend sind ja die Trainingsdaten. Der gleiche Algorithmus wird dieselben Daten ja anders bewerten, wenn er anders trainiert wurde – und so kann es einer weiteren Arbeitsgruppe unmöglich sein, Ergebnisse einer Publikation nachzuvollziehen.
Falk zeigt Verständnis für diese Sorge und bestätigt, dass man auch in U-Net einen Bias hineintrainieren kann. Doch diesen Bias habe man ja ohnehin immer gehabt, in Zeiten als der Mensch noch selber am Mikroskop zählte. Immerhin lasse sich das Problem jetzt ein Stück weit in den Griff bekommen, wenn unterschiedliche Trainingsdaten von mehreren erfahrenen Forschern annotiert sind.
Einmal trainiert ist ein Tool wie U-Net aber unbestechlich, hebt Falk hervor: „Der Algorithmus wird dann jeden Datensatz gleich behandeln. Zudem wird er auch nicht müde und wendet tags wie nachts dieselben Kriterien an, wodurch die Ergebnisse dann doch wieder vergleichbar sind.“
U-Net gehört zu den KIs, die gewissermaßen unter Anleitung trainieren – Informatiker sprechen von „überwachtem Lernen“. Aus den Trainingsdaten bekommt die Software ein direktes Feedback, ob ein jeweiliges Detail korrekt oder fehlerhaft kategorisiert oder quantifiziert worden ist. Es gibt aber auch KIs, die sozusagen ohne Trainer auskommen. Sie suchen eigenständig nach Mustern. Forscher vom Helmholtz Zentrum München haben solch ein Tool programmiert, um Fehler in Datensätzen aus Einzelzell-Transkriptomen zu korrigieren. Ihren Deep Count Autoencoder (DCA) haben die Entwickler im Januar in Nature Communications vorgestellt (10(1): 390).
Einer von ihnen ist Lukas Simon. Der Bioinformatiker forscht in der Arbeitsgruppe von Fabian Theis und interessiert sich speziell für die Analyse von Einzelzell-Daten. Will man die Transkriptome einzelner Zellen bestimmen, so sollte im Idealfall jede einzelne mRNA einer Zelle erfasst werden. Natürlich wird man dieses Ideal nie erreichen. Doch was ist, wenn man in Transkriptom-Daten auf eine Zellpopulation stößt, die von einem bestimmten Gen X überhaupt nichts exprimiert hat? Wenn dort also für Transkript X immer eine „Null“ steht?
„Es gibt zwei Arten von Nullen“, veranschaulicht Simon. „Bei den biologischen Nullen ist tatsächlich keine mRNA in der Zelle exprimiert gewesen. Daneben gibt es jedoch noch technische Nullen – und die wollen wir korrigieren“. Bei einer „technischen Null“ ist die fehlende Sequenz ursprünglich sehr wohl als RNA-Molekül durch die Zelle geschwommen, doch aus irgendeinem Grund wurde sie nicht erfasst. „Man diskutiert unterschiedliche Ursachen dafür“, erläutert Simon. „Zum Beispiel kann die RNA schon abgebaut gewesen sein, bevor sie in der Reaktionskammer für die reverse Transkription eingefangen werden konnte.“ Solche „falschen Nuller“ nennt man Dropouts.
Komprimieren über Muster
Die Idee ist nun, dass ein Algorithmus in den Daten Muster erkennt. So sollten ähnliche Zelltypen oder Zellen in ähnlichen Stadien Gemeinsamkeiten in ihren Transkripten aufweisen. Sie bilden Cluster, und so kann man in den Daten Gruppen und Subgruppen ausfindig machen. Bleiben wir wieder bei unserem hypothetischen Transkript von Gen X: Landen alle Nullwerte auch im Hinblick auf andere Merkmale in gemeinsamen Clustern, so dürfte es sich um biologische Nullen handeln. Sind sie mehr oder weniger zufällig durch alle möglichen Zellpopulationen verteilt, in denen X sonst hochreguliert ist, sind es wohl Dropouts. „Das sieht ein Mensch eigentlich auch sofort in den Daten“, meint Simon – nur sequenziert man mitunter Millionen von Zellen pro Durchlauf. Der Mensch ist also definitiv überfordert, wenn er von Hand nach Dropouts suchen soll.
Zunächst scheint es grotesk, dass ein Algorithmus völlig untrainiert Dropouts erkennen kann. DCA gehört nun aber zu einer gängigen Klasse neuronaler Netze: eben den sogenannten Autoencodern. „Ein Autoencoder hat eine Input-Schicht, die genauso groß ist wie die Output-Schicht; und er soll einen Output erzeugen, der dem Input möglichst ähnlich ist“, geht Simon auf die programmtechnischen Details ein. Anschaulicher kann man sich als Eingabe eine riesige Tabelle mit Werten vorstellen. Um die Werte dieser Tabelle weiterzuverarbeiten, muss der Autoencoder in Zwischenschritten deutlich kleinere Tabellen verwenden – um am Ende die Ursprungstabelle wieder möglichst genau zu rekonstruieren. „Mit anderen Worten: Ein Autoencoder lernt, Daten zu komprimieren und ist damit gezwungen, ein Muster in den Daten zu erkennen, anstatt einzelne Datenpunkte auswendig zu lernen.“
Finde die falsche Null
Simon nennt ein simples Beispiel: „Sie haben eine Tabelle mit 100.000 Zeilen und 100.000 Spalten. In jeder Zelle steht eine Null, außer in den Zellen, die die Diagonale bilden – dort steht jeweils eine Eins. Sie hätten jetzt keine Probleme, diese Tabelle aus dem Kopf heraus wieder herzustellen; Sie merken sich nicht jedes einzelne Element sondern nur das Datenmuster.“ Entsprechend sucht auch der DCA nach Mustern in den Daten. „Der Autoencoder gleicht dann die Input-Daten mit dem erlernten Datenmuster ab, um die Wahrscheinlichkeit zu berechnen, dass es sich bei einer Null um ein Dropout-Artefakt handelt“, fährt Simon fort. In diesem Fall ersetzt der DCA die Null durch einen plausibleren Wert.
Natürlich muss sich der Anwender auch hier über die Grenzen seines Tools im Klaren sein. „Wenn ein Transkript ohnehin niedrig exprimiert ist, fällt es dem DCA schwerer, Dropouts von biologischen Nullen zu unterscheiden.“ Außerdem dürfe man auch nicht erwarten, aus einem sehr verrauschten Datensatz plötzlich tiefe Erkenntnisse zu gewinnen. „Die Grundstruktur der Daten wird sich nicht ändern“, betont Simon. „Wenn sich ein Biologe aber für den Vergleich zweier Gene interessiert, dann ist der DCA ein hilfreiches Werkzeug.“
Auch wenn uns meist DNA und RNA als Erstes einfallen, wenn wir an Big Data in den Lebenswissenschaften denken: Was am Ende in der Zelle tatsächlich passiert, das ist maßgeblich Sache der Proteine. Und die steuern wiederum Reaktionsflüsse kleinerer Moleküle innerhalb der Zelle. Eine wesentliche Kennzahl ist hier der kcat-Wert. „Der Wert gibt an, wie viel Substrat ein aktives Zentrum pro Sekunde maximal umsetzen kann“, erläutert der Bioinformatiker David Heckmann. Hat ein Protein einen kcat von 10, so kann es zehn Substratmoleküle pro Sekunde reagieren lassen, falls es mit Substrat vollständig gesättigt ist. Mittels Machine Learning haben Heckmann und einige Koautoren nun ein Modell entwickelt, um die kcat-Werte von Proteinen vorherzusagen (Nat. Commun. 9(1): 5252).
Bis 2016 arbeitete Heckmann als Postdoc an der Uni Düsseldorf in der Gruppe von Martin Lercher, der ebenfalls am genannten Paper mitgeschrieben hat. Heute ist Heckmann in La Jolla an der University of California und integriert Metabolom- und Proteom-Daten in Computermodelle. Im aktuellen Projekt bestand eine wesentliche Herausforderung darin, biochemische Daten aus Datenbanken und Veröffentlichungen bis zurück in die 80er Jahre zu sammeln und in eine verwertbare Form zu überführen. „Da kann man nicht gerade sagen, dass die Daten standardisiert waren“, erinnert er sich an den Aufwand bei der Datenaufbereitung.
Bislang haben Heckmann und Kollegen ausschließlich E. coli berücksichtigt – einfach weil von diesem Organismus die meisten kinetischen Daten zur Verfügung stehen. Heckmann hofft aber, dass man ähnliche Modelle künftig auch für andere Organismen etablieren und für praktische Anwendungen nutzen kann. „Den kcat können Sie sich prinzipiell vorstellen als die Kosten, die eine biochemische Reaktion fordert“, erklärt er. Katalysiert ein Enzym mit hohem kcat einen Stoffumsatz, so ist diese Reaktion „kostengünstiger“, weil weniger Enzym synthetisiert werden muss. „Das könnte zum Beispiel interessant sein, wenn wir die Düngerverwendung optimieren oder neue Pflanzenformen züchten wollen“, nennt Heckmann Beispiele für mögliche Anwendungen. Man könnte dann nämlich im Vorfeld überlegen, welche Stoffwechselwege man vorzugsweise ansteuert.
Die Beispiele zeigen, dass in den Lebenswissenschaften vielseitige Anwendungsmöglichkeiten für die Entwickler von KI-Systemen warten. Wünschenswert ist natürlich, dass der Mensch die Voraussagen seiner Computermodelle auch nachvollziehen kann. Doch bei sehr komplexen Zusammenhängen sind wir vielleicht auch froh, wenn uns der Computer da weiterhilft, wo unser Denken versagt. Gerade in der Medizin, wo Algorithmen hoffentlich bald die Diagnostik verbessern und dabei helfen, die optimale Therapie auszuwählen.
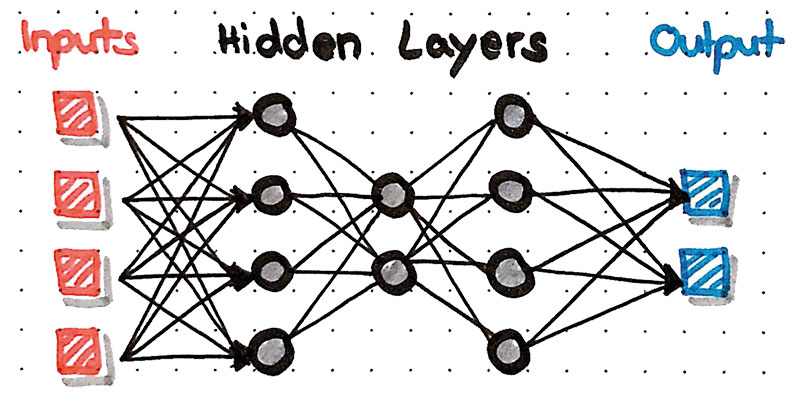
Was ist eigentlich ein künstliches neuronales Netz?
Künstliche Intelligenz (KI) und Deep Learning klingen modern, die Grundkonzepte dahinter sind aber nicht neu. Viele moderne Methoden greifen auf jahrzehntealte Ideen aus der Informatik zurück. So etwa die künstlichen neuronalen Netze, die sich tatsächlich an ihren biologischen Pendants orientieren – nur dass sie eben am Computer mathematisch dargestellt werden. Klassischerweise arrangiert man die künstlichen Neuronen in mehreren Schichten – dabei beeinflusst die erste Schicht die zweite, diese dann die dritte und so weiter. Die erste Schicht entspricht den eingegebenen Daten, die letzte Schicht repräsentiert die Ausgabe des Ergebnisses. Das bedeutet: Wenn etwas verrechnet werden soll, so muss solch ein neuronales Netz aus mindestens zwei Schichten bestehen. Weiterhin kann jedes Neuron im Netzwerk einen bestimmten Wert annehmen, sagen wir eine Dezimalzahl zwischen Null und Eins.
Ein Beispiel: Angenommen, unser neuronales Netz soll auf Schwarzweißfotos abgebildete pflanzliche Lebensmittel einteilen in die Kategorie „Obst“ oder „Gemüse“. Jedes Foto hat eine Auflösung von 200 mal 200 Pixeln, wird also von insgesamt 40.000 Bildpunkten repräsentiert. Unsere erste Schicht bestünde somit aus 40.000 Neuronen – eines für jeden Bildpunkt. Nun hat jedes Pixel einen Grauwert – den können wir umwandeln in einen Wert zwischen Null und Eins. Damit ist die erste Schicht unseres Netzwerks zunächst einmal langweilig, denn sie kodiert ja einfach nur das Foto.
Jedes Neuron der ersten Schicht hat aber eine Verbindung mit jedem Neuron der zweiten Schicht – ähnlich wie biologische Neurone durch Synapsen verbunden sind. Jede dieser Verbindung hat eine bestimmte Gewichtung, mit welcher der Wert des Neurons der ersten Schicht weitergegeben wird an die zweite Schicht. Diese Gewichtung kann zudem positiv oder negativ sein – ähnlich wie wir aus der Biologie hemmende und aktivierende Synapsen kennen. In der zweiten Schicht wird nun für jedes Neuron ein Wert errechnet. Dazu werden alle Eingänge, die dieses Neuron bekommt, zunächst einmal aufaddiert und anschließend über eine definierte Regel (eine mathematische Funktion) wieder zu einem Wert zwischen Null und Eins normiert.
Nun beginnt dasselbe Spiel von vorn, nur dass „Signale“ von der zweiten Schicht zur dritten Schicht weitergereicht und verrechnet werden. Am Ende gibt die letzte Schicht dann ein Ergebnis aus. In unserem Fall sind das zwei Neurone, die auch wieder jeweils Werte von Null bis Eins annehmen können. Es gibt ein „Obst-Neuron“ und ein „Gemüse-Neuron“.
„Zeigt“ man dem Netzwerk nun das Foto eines Apfels, so sollte im Idealfall ein „Obst-Wert“ von Eins und ein „Gemüse-Wert“ von Null herauskommen. Genau andersherum wäre es bei einer Karottenwurzel. Worauf kommt es nun an, ob unser „Obst-Gemüse-Kategorisierer“ gut oder schlecht funktioniert? Offenbar sind es die Gewichtungen der Verbindungen zwischen den Neuronen, die die Verrechnung bestimmen, sowie die Anzahl der Neurone in den zwischenliegenden Schichten – und die Anzahl der Schichten. Wir starten ja mit einer Unmenge von 40.000 Neuronen und sehen sofort: Eine sinnvolle Konfiguration „von Hand“ ist nicht möglich. (Mal ganz davon abgesehen, dass es mitunter schon Menschen schwerfällt, Obst und Gemüse zu definieren).
Man probiert also erstmal eine bestimmte Architektur aus – legt eine Anzahl von Schichten zwischen der Eingabe- und Ausgabeschicht fest (diese Zwischenschichten heißen Hidden Layers) und wählt für jede Zwischenschicht eine Anzahl von Neuronen. Die Gewichtungen der Verbindungen legen wir zunächst zufällig fest. Nun geben wir dem Netzwerk etliche Fotos, auf denen entweder ein Obst oder ein Gemüse abgebildet ist. Das Netzwerk lernt, indem es seine Ausgabe mit derjenigen Ausgabe vergleicht, die korrekt wäre. Während des Trainings ist also ein Feedback notwendig, wie nah das ermittelte Ergebnis an dem gewünschten Ergebnis lag.
Im Training verändert das Netzwerk die Verbindungen zwischen den Neuronen in jedem Schritt leicht ab – und zwar immer in eine Richtung, die die Ergebnisse bei Verarbeitung der Trainingsdaten verbessern. Verbindungen, die zu einer unerwünschten Aktivität in den Ausgabeneuronen führen, werden dabei ein bisschen geschwächt – andere verstärkt. Mathematisch betrachtet haben wir es mit einer sehr unhandlichen Funktion zu tun, in der jede Neuronenschicht einem Vektor mit etlichen Komponenten entspricht. Und beim Training sucht das Netzwerk dann nach einem lokalen Minimum dieser Funktion.
Irgendwann wird das Netz so konfiguriert sein, dass es die Trainingsdaten zuverlässig erkennt. Nun wird es spannend: Kategorisiert der Algorithmus auch Bilder korrekt, die ihm bislang unbekannt waren? Falls nein, sollten wir das nicht so schwer nehmen, denn die Definitionen für Obst und Gemüse sind ohnehin nicht immer ganz einheitlich. Funktioniert das Tool aber, so sollten wir uns nicht zu früh freuen: Zeigen wir dem Netzwerk jetzt doch mal das Foto eines Blauwals, einer Schreibmaschine oder einer Spiralgalaxie! Gut möglich, dass auch diese Bilder ganz klar als entweder „Obst“ oder „Gemüse“ einsortiert werden. Natürlich sind diese Ergebnisse unsinnig, doch unser neuronales Netz hat ja nun mal nichts anderes gelernt, als sich bei einem eingegebenen Bild möglichst klar für „Obst“ oder „Gemüse“ zu entscheiden.
Insofern ist es also wichtig, die Grenzen seiner KI zu kennen beziehungsweise diese erst einmal sorgfältig auszutesten. Man sollte sich immer im Klaren sein, für welche Aufgabe ein System trainiert ist. In diesem Fall können wir bestenfalls Fotos zuverlässig in die richtige Schublade sortieren, von denen wir bereits wissen, dass sie garantiert nichts anderes zeigen als entweder ein Obst oder ein Gemüse. Wollen wir hingegen aus einer komplexeren Bildersammlung nur die Fotos filtern und sortieren, auf denen Obst oder Gemüse zu sehen ist, müssen wir unser Tool anders trainieren.
Letzte Änderungen: 29.11.2019