Auflösung durch Abregung
Am 23. November hat Bundespräsident Horst Köhler den mit 250.000 € dotierten 10. Deutschen Zukunftspreis verliehen.

(24.11.2006) Der 10. Deutsche Zukunftspreis geht an
Stefan W. Hell (Photo rechts) vom Max-Planck-Institut für Biophysikalische Chemie, Göttingen für sein Durchbrechen der Abbeschen Beugungsgrenze und die daraus folgende Erfindung des
Stimulated Emission Depletion Microscope, kurz STED-Mikroskop.
Nein, die Jenaer müssen
Ernst Karl Abbe (Photo links) nicht von seinem Sockel holen, denn falsch ist seine Formel zur Berechnung der Auflösungsgrenze von Lichtmikroskopen nicht:
d
min = λ / (n * sin (α
/2)
d
min ist hier der Mindestabstand zweier Punkte, die gerade noch aufgelöst werden, bessere Auflösung heißt kleineres d
min. Und verringern kann man d
min durch:
1. Vergrößern von α
/2. Dies ist der halbe Öffnungswinkel des Objektivs, kann im Mikroskop nicht größer werden als 70 Grad.
2. Vergrößern des Brechnugsindex n. Bei Immersionsobjektiven liegt der Wert bestenfalls bei ca. 1.4.
3. Verringerung der Wellenlänge λ. Verwendet man Blaulicht, kommt man auf ein λ von 400 nm.
Also berechnet sich die Abbesche Grenze der Auflösung von Lichtmikroskopen auf zirka 200 nm. Dies gilt für die Verwendung sichtbaren Lichts und eine gleichmäßige Durchstrahlung des Objektes im Weitfeld.
Ernst Ruska durchbrach als erster die Abbesche Grenze, als er in 30er Jahren des letzten Jahrhunderts statt Licht Elektronen mit deutlich geringerer Wellenlänge verwendete. Das Elektronenmikroskop war geboren. Mit ihm kann man d
min auf zirka 0,1 nm verringern. Allerdings liegt im Elektronenmikroskop das Objekt im Vakuum unter Elektronenbeschuss - lebende Zellen kann man so nicht mikroskopieren. Aber natürlich gebührt Ernst Ruska ein Sockel neben Ernst Abbe.
Die jetzt durch Stefan Hell eingeleitete Revolution in der Lichmikroskopie ist sicherlich an Bedeutung mit der Entdeckung des Elektronenmikroskops zu vergleichen und Stockholm sollte sich schon mal einen Termin vormerken. Denn Stefan Hell hat d
min auf zirka 20 nm reduziert - wohlgemerkt im Fluoreszenzmikroskop, einem Lichtmikroskop, und damit auch bei lebenden Zellen anwendbar.
Wie soll das funktionieren - denn auch das von einem fluoreszierenden Molekül ausgesandte Licht wird gebeugt und der Beugungsfleck kann nicht kleiner werden als 200 nm?
Mit einem Trick! Hier kommt die Abregung ins Spiel. Denn man kann mit Licht ein Molekül nicht nur zur Fluoreszenz anregen, sondern auch sofort abregen.
Bei der Fluoreszenz wird ein Molekül durch einen Lichtstrahl einer bestimmten Wellenlänge angeregt und gibt die erhaltene Energie spontan wieder als Licht einer anderer Wellenlänge, nämlich der Fluoreszenzwellenlänge, ab.
Bei der Abregung wird ein vorher angeregtes Molekül mit einem weiteren Lichtstrahl bestrahlt. Das Molekül gibt dann die durch die Anregung erhaltene Energie nicht als Licht der Fluoreszenzwellenlänge ab, sondern als Licht der Abregungswellenlänge. Dies wird stimulierte Emission genannt.
Wählt man nun für die Abregung eine andere Wellenlänge als die Fluoreszenzwellenlänge, so kann man das Licht aus Fluoreszenz und Abregung gut voneinander unterscheiden.
In der Regel hat man es aber mit mehr als einem angeregten Molekül zu tun. Daher ist es wichtig, dass die Abregung übersättigt werden kann: Ab einer gewissen Intensität des Abregungsstrahls ist die verbleibende Population von Molekülen im angeregten Zustand vernachlässigbar gering, eine quasi komplette Abregung ist möglich. Und das Beste ist: Der Prozess der Abregung ist reversibel, abgeregte Moleküle können immer wieder angeregt und abgeregt werden.
Das ist sicherlich alles sehr schön bunt, aber wie wird damit die Auflösung verbessert?
Lassen Sie uns ein Objekt mit einem Lichtstrahl Punkt für Punkt rastern. Sie werden erneut einwenden, durch Beugung ist der Lichtpunkt, mit dem wir das Objekt rastern, mindestens 200 nm groß und löst mithin nicht besser auf.
Stefan Hell rastert das Objekt mit dem STED-Mikroskop deutlich raffinierter ab. Dessen Anregungsstrahl hat einen Durchmesser von zirka 250 nm. Darüber legt er einen Abregungsstrahl, der den Anregungsstrahl überdeckt, aber in der Mitte über eine Nullstelle verfügt - es ist also genau gesagt ein Abregungs-Ring, in der Mitte wird nicht abgeregt. So empfängt Stefan Hell nur Fluoreszenzsignale aus der Mitte des Abregungsringes. Und damit hat er den Bereich, aus dem er Lichtsignale erhält, deutlich unter 200 nm gedrückt. Den Abregungsring erzeugt er übrigens mit einer Phasenplatte. Und je höher die Intensität des Abregungslichtes ist, umso kleiner ist das Loch in der Mitte des Abregungsringes und umso höher ist die Auflösung des Bildes.
Zur Veranschaulichung nun das Ganze an einem Beispiel in Farbe:
1. Fall: Ein Präparat wird mit einem Spot UV-Licht von 200 nm Durchmesser angeregt. Die Fluoreszenz strahlt grün, in einem Spot von 200 nm.
2. Fall: Das Präparat wird mit einem Spot UV-Licht von 200 nm Durchmesser angeregt. Gleichzeitig wird mit einem Rotlicht Spot von 200 nm durchstrahlt. Die Probe leuchtet rot, in einem Spot von 200 nm. Das ist die stimulierte Emission.
3. Fall: Das Präparat wird mit einem Spot UV-Licht von 200 nm Durchmesser angeregt. Gleichzeitig wird mit einem Rotlicht Ring durchstrahlt. Nur in der Mitte des 200 nm Spots sieht man die grüne Fluoreszenz - der Rest des Spots leuchtet rot.
In einer Abbildung von Stefan Hell selbst sieht das so aus:
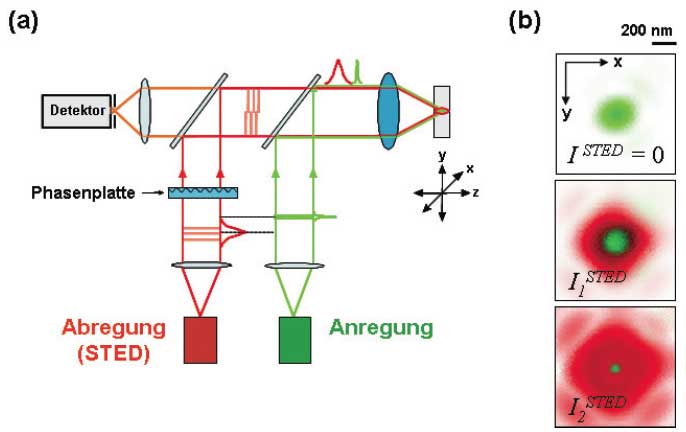
Fig. 1: STED-Mikroskopie: (a) Typisches Schema eines STED-Mikroskops mit Anregungs- und Abregungsstrahl, Phasenplatte, Detektor und Objektiv. In der Fokalebene (x,y) des Objektivs bildet der Anregungslichtpunkt (grün) eine Scheibe mit zirka 250 nm Durchmesser aus (b, rechts oben), die mit dem zeitlich synchronisierten Abregungslichtpuls überlappt (b, rechts mittig). Die Phasenplatte bildet den stimulierenden Abregestrahl so um, dass er nicht ebenfalls eine Scheibe, sondern eine zentrale Nullstelle ausbildet. Das Abregungslicht wird bei niedriger STED-Sättigung nur im äußeren Bereich des Anregungsfokus wirksam. Mit zunehmender Intensität bleibt jedoch nur ein immer kleinerer Bereich von der Abregung durch stimulierte Emission ausgenommen (b, rechts unten), so dass der Fokalbereich, in dem Fluoreszenz noch möglich ist, also der Fluoreszenzspot, weit unter die Beugungsgrenze gedrückt wird. (a) Abrastern in x,y eines fluoreszenzmarkierten Objekts mit diesem kleinen Spot liefert Bilder mit Auflösungen weit unterhalb der Beugungsgrenze.
Prinzipiell ist die Auflösung des STED-Mikroskops also abhängig von der Stärke der Überstrahlung und der daraus resultierenden Größe der zentralen Nullstelle des Abregungsspots. In der biologischen Anwendung stellt sich noch die Frage, wie groß der Energieeintrag durch Abregungslicht ist, den eine Probe verträgt.
Alles in allem aber ist das Verfahren von Stefan Hell so elegant und überzeugend, dass die deutsche Forschungsförderung Stefan Hell schon früh unterstützte und er dieses Konzept in einem bestens ausgestatteten Labor mit eigener Arbeitsgruppe entwickeln konnte.
Nun ja, so sehr der erste Teil des obigen Satzes gilt, so sehr gehört der zweite Teil doch in die Welt der Sagen und Märchen.
Schon während seiner Doktorarbeit Ende der 80er Jahre fing Hell an, an der Abbeschen Grenze zu knabbern. Sein erstes Projekt, die 4Pi-Mikroskopie entwickelte und patentierte er nach seiner Promotion als freier Erfinder.
Mit dieser Erfindung erhielt er immerhin ein Postdoc-Stipendium der DFG am EMBL in Heidelberg. Leider hatte er dort aber weder ein eigenes Labor noch akademische Förderung.
Es war ein finnischer Kollege am EMBL, der Hell seinem Professor in Turku, Finnland, vorstellte. Dort erhielt Hell Freiraum für seine Arbeit und schließlich auch ein Stipendium der Finnischen Akademie. Aber auch in Finnland wurde das Geld knapp und die Lizenzierung des privaten 4Pi-Mikroskopie-Patents musste zur Finanzierung herangezogen werden.
Als dann schließlich der damalige geschäftsführende Direktor des Göttinger MPIs für Biophysikalische Chemie auf Hells Forschung aufmerksam wurde, erhielt er dort eine Fünf-Jahres-Stelle als Leiter einer Max-Planck-Nachwuchsgruppe, die er mit zusätzlichen Mitteln des BMBF weiter ausbauen konnte.
Zur Ehrenrettung der deutschen Forschungsförderung sei aber die Patentstelle der Fraunhofer-Gesellschaft erwähnt, die noch während Hells Zeit in Finnland die Patentanmeldung des STED-Mikroskops finanziell unterstützt hat.
Mittlerweile steht das STED-Mikroskop kurz vor seiner Markteinführung durch die Firma Leica Microsystems in Wetzlar, einer Tochter der US-amerikanischen Danaher Corporation.
Genaue Angaben zu Verfügbarkeit, Preis undsoweiter konnte uns Leica Microsystems noch nicht mitteilen. Wir bleiben aber am Ball und werden weiter über die STED-Mikroskopie berichten.
Als Ausblick zeigen wir eine STED-Aufnahme aus dem Labor Hell. Durch die Komprimierung für die Webdarstellung hat die STED-Aufnahme leider etwas gelitten - im Druck ist die Aufnahme noch deutlich beeindruckender.
Fig. 2: Bild mit Schärfe jenseits der Auflösungsgrenze. Der Vergleich zeigt die Verteilung des Proteins Syntaxin1A in der Membran einer Säugerzelle mit normaler Auflösung (links) und mit Überauflösung durch STED (rechts). Syntaxin liegt nicht-homogen verteilt in der Membran vor. Es spielt eine zentrale Rolle bei der Fusion von Vesikeln mit der Plasmamembran, deren besseres Verständnis einen genaueren Einblick in den Ablauf intrazellulärer Transportprozesse erlauben wird. Die durch STED erzielte Auflösung von 75 nm ermöglicht eine viel aussagekräftigere Erfassung der lokalen Verteilung von Syntaxin-Clustern.
Abschließend möchten wir uns bei Herrn Stefan W. Hell und der Pressestelle des MPI für Biophysikalische Chemie in Göttingen für die Möglichkeit bedanken, zwei Abbildungen in diesen Artikel zu übernehmen. Die Abbildungen stammen aus:
Hell, Stefan W. Neues Gesetz zur Auflösung in der Lichtmikroskopie ermöglicht Bilder in bisher ungekannter Schärfe.
Jahrbuch der Max-Planck-Gesellschaft 2005.
Carsten T. Rees
Letzte Änderungen: 11.12.2006