Auf in neue Dimensionen!
cAMP-Zauberstab und 3D-Gelektrophorese
Hubert Rehm
Eine bewährte Methode zu verbessern ist schwer genug dabei in neue Dimensionen vorzustoßen, ist jedoch eine Glanzleistung, die selbst Hubert Rehm begeistert.
Wer intrazelluläre Signalwege untersucht, wünscht sich oft einen Zauberstab. Damit könnte er blitzschnell eine definierte Menge eines intrazellulären Botenstoffs ins Cytosol zaubern und messen, wie die Zelle darauf reagiert. Am besten wäre ein Stab für alle sekundären Signalträger, für cAMP, Ca
2+, cGMP, IP3 und NO, doch wäre der Forscher schon zufrieden, wenn sein Zauberstab wenigstens über einen bestimmten Botenstoff Macht hätte, am liebsten über den wichtigsten, das cAMP. Denn cAMP reguliert intrazelluläre Abläufe fast aller Organismen - von der Alge bis zum Menschen. Über die Aktivierung von Proteinkinase A bestimmt es die Expression von Genen oder setzt aus Glykogen Glukose frei.
Eine Methode, die cAMP-Konzentration in der Zelle schlagartig zu erhöhen, ist das "photouncaging". Entwickelt wurde sie Mitte der 70er Jahre von dem Habilitanden Joachim Engels. Seine Idee war, Zyklophosphate als Benzylester zu maskieren und sie in der Zelle durch Hydrolyse oder Photolyse freizusetzen. Schon 1977 publizierte Engels (
J. Med. Chem, 20, 907) die Arbeit "Synthesis, Structure and Reactivity of Adenosine Cyclic 3',5'-phosphate Benzyl Triesters" und 1978 eine Arbeit über caged-cGMP (
Experientia 1978, 34, 14).
Bewährt aber nicht optimal
Die Anwendung des caged-cAMP ist einfach. Zu den Zellen wird 4,5-dimethoxy-2-nitrobenzyl-cAMP (DMNB-cAMP) gegeben. Die Verbindung ist membrangängig und erreicht daher das Zytosol. Dort setzt einige Sekunden Bestrahlung mit UV-Licht das cAMP frei. Jedoch hat die Methode Nachteile: niedrige Löslichkeit in wässrigen Puffern, niedrige Quantenausbeuten bei der Photolyse, geringe Photolysegeschwindigkeit, Instabilität in wässriger Lösung. Man hat daher andere Verbindungen entwickelt, zum Beispiel das Azyloxy-Cumarinylmethyl-caged-cAMP. Aber auch diese Verbindungen zeigten Nachteile und für alle gilt: ihr Reservoir ist endlich. Ist die Verbindung vollständig zerfallen, kann kein cAMP mehr produziert werden, die Zellen müssen - wenn sie die Bestrahlung überlebt haben - erneut mit caged-cAMP inkubiert werden. Zudem kann man nur mit Zellen in Kultur arbeiten.
Die Gruppe von Georg Nagel am Max-Planck-Institut für Biophysik hat im Januar dieses Jahres in
Nature Methods (4, 39-42) eine Methode vorgestellt, die einem cAMP-Zauberstab noch näher kommt.
Auf Tümpeln und Wasserlachen schwimmt im Sommer oft ein grüner Film. Er besteht zum größten Teil aus Algen wie dem Einzeller
Euglena gracilis, auch Augentierchen genannt.
Euglena besitzt Geißeln, Chloroplasten und eine photoaktivierbare Adenylatzyklase. Die Zyklase ist ein Tetramer aus zwei α- und zwei β-Einheiten (α-PAZ, 112 kDa und β-PAZ, 94 KDa). Jede der Einheiten besitzt Zyklaseaktivität. Sie wird durch Blaulicht erhöht, denn das Enzym ist Teil einer Signalkette, die es der Alge ermöglicht, sich zu photosynthetisch nutzbarem Licht hin beziehungsweise von schädlichem UV-Licht weg zu geißeln.
Die Doktorandin Saskia Schröder-Lang und der Emmy-Noether-Stipendiat Martin Schwärzel haben sowohl α-PAZ als auch β-PAZ in Xenopus-Oozyten, HEK293-Zellen (eine menschliche embryonale Nierenzelllinie) und Drosophila exprimiert.
Die cAMP-Konzentration in unstimulierten normalen (Kontroll)-Oozyten (totales cAMP = gebundenes und freies) liegt bei 1-2 mM. Wenn in den Oozyten α-PAZ exprimiert wurde (mit 3 ng cRNA) und man die Oozyten im Dunkeln hielt, lag die totale cAMP-Konzentration etwa 20 mal höher. Offensichtlich ist α-PAZ auch ohne Bestrahlung aktiv. Eine fünfminütige Be-strahlung mit blauem Licht jedoch steigert die cAMP-Konzentration noch einmal um das zehnfache. Wurde dagegen β-PAZ exprimiert, blieb die cAMP-Konzentration die gleiche wie bei Kontrollzellen, gleich ob bestrahlt wurde oder nicht.
Bestimmt wurde die totale cAMP-Konzentration, interessant wäre jedoch ein Nachweis des freien cAMP in der Zelle. Dazu nutzten Schröder-Lang et al. einen menschlichen Chloridkanal, den Transmembranleitfähigkeitsregulator von zystischer Fibrose (hTLRZF), denn zystische Fibrose ist ein weiteres Interessengebiet von Nagel. Der Kanal wird durch cAMP abhängige Proteinkinase aktiviert. Die freie cAMP-Konzentration im Zytosol kann so indirekt über die Membranleitfähigkeit gemessen werden.
Blaulicht öffnet Kanäle
In die Oozyten wurden α-PAZ-cRNA (wegen der hohen Dunkelaktivität nur 200 pg) und hTLRZF-cRNA injiziert. Das Ergebnis: Ein Blaulichtpuls von fünf Sekunden erhöhte mit einer Verzögerung von 15 bis 20 Sekunden die Membranleitfähigkeit der Oozyten. Mit 200 pg β-PAZ-cRNA änderte sich die Membranleitfähigkeit nicht. Erst bei einer 100-fach höheren Menge von β-PAZ-cRNA war eine Änderung messbar.Eine direkte Messung der freien cAMP-Konzentration ist mit zyklische-Nukleotid-gesteuerten Kanälen möglich, denn diese Kanäle werden direkt durch cAMP geöffnet. Es handelt sich um unspezifische Kationenkanäle, die unter anderem für Ca
2+ permeabel sind. Die Koexpression von α-PAZ und einem cAMP-abhängigen Kanal (halbmaximale Aktivierung bei 14 mM cAMP) ergab erwartungsgemäß eine wesentlich schnellere Änderung der Membranleitfähigkeit. Experimente mit HEK293-Zellen, hier wurde der Ca
2+-Einstrom mit Fluo-4 gemessen, ergaben ähnliche Ergebnisse.
Mit diesen schönen Ergebnissen hätten es andere bewenden lassen. Es wäre dann ein solide Methode gewesen, mit dem Vorteil, dass man in einer Zelle die cAMP- Konzentration so oft variieren kann wie man Lust hat und die Zelle das Blaulicht verträgt. Das Ausgangsprodukt für cAMP, das ATP, steht ja unbegrenzt zur Verfügung. Man hätte die Methode auch in einer soliden Zeitschrift publizieren können und die Fachwelt hätte gesagt: "Schön und gut, aber ach, da muss ich ja im Dunkeln munkeln und, du liebes bißchen, da muss ich ja cRNA herstellen. Da greif ich doch lieber zum caged-cAMP."
Martin Schwärzel arbeitet jedoch an der Universität des Saarlandes am Gedächtnis von
Drosophila und da schien sich eine weitere Anwendung der Methode anzubieten. Schwärzel exprimierte mit dem Gal4-UAS-Verstärker-System die PAZ-Enzyme im Fliegenhirn. Erwachsene Fliegen schien das nicht zu beeindrucken, sie süffelten weiterhin ruhig ihren Apfelsaft. Bei blauem Licht jedoch wurden die α-PAZ-Fliegen unruhig und zeigten ungewöhnliches Frierverhalten. Zudem änderte sich das Putzverhalten. Werden Fruchtfliegen mit Pulver bestreut, fangen sie sofort und mit Ausdauer an, sich zu putzen. Bestrahlt man pulverbestreute α-PAZ-Fliegen mit blauem Licht, stellen sie die Putzerei ein, während Wildtypfliegen nach wie vor versuchen, das Pulver los zu werden. Schaltet man das Licht ab, fangen die α-PAZ-Fliegen wieder mit dem Putzen an. Sie können sich das im Netz in einem Film anschauen:
http://www.nature.com/nmeth/journal/vaop/ncurrent/suppinfo/nmeth975_S1.html
Alle Experimente wurden sauber kontrolliert, die Effekte sind eindrucksvoll und eindeutig.
Die Variation von cAMP nach Belieben am lebenden Ganztier, das ist sensationell, das ist mit caged-cAMP nicht möglich, diese Technik ermöglicht Experimente von denen man vorher nur träumen konnte. So könnte man mit einem fokussierten Laserstrahl in ganz bestimmten Neuronen zu ganz bestimmten Zeiten das cAMP erhöhen und dann die Auswirkungen auf das Verhalten beobachten. Mit den Worten von Schröder-Lang et al.: "This tool may provide exquisite spatiotemporal control of cAMP levels in future work of signaling pathways in transgenic models, particularly for tackling questions of learning and memory in Drosophila." Die cAMP-Variation hat zwei neue Dimensionen gewonnen: Eine im Raum und eine in der Zeit.
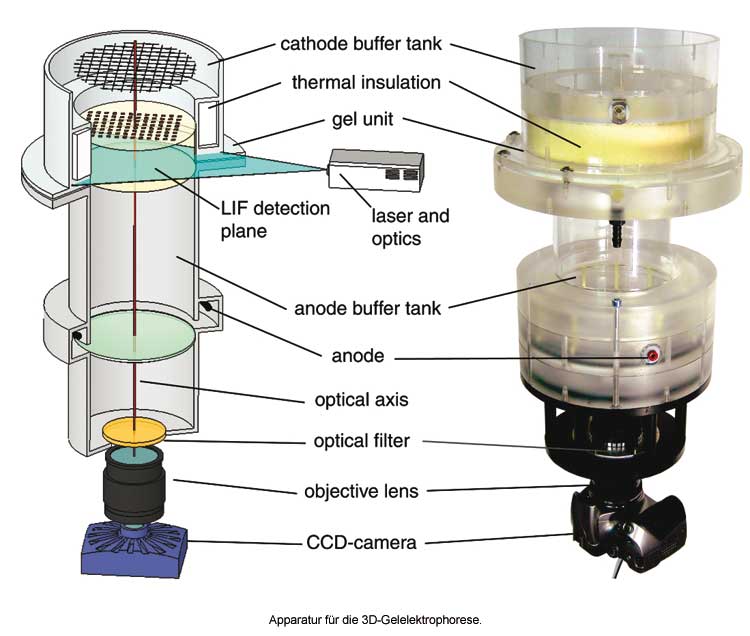
Nur eine neue Dimension, aber immerhin, haben Robert Ventzki et al. für die altbekannte Gel-Elektrophorese gewonnen (Biotechniques 2007, 42(3), 271-279). Bekanntlich gibt es die eindimensionale (1D) und die zweidimensionale (2D) Gelelektrophorese. Ventzki et al. haben eine 3D-Elektrophorese entwickelt. Als Dritte Dimension dienen Zeit, Konzentration oder ein drittes Trennprinzip. Letzeres könnte nach folgendem Prozedere ablaufen: Man trennt eine Proteinmischung mit der Blaugel-Elektrophorese auf. In zweiter Dimension wird das Blaugel auf ein Slab-Gel gelegt und isoelektrisch fokussiert. In dritter Dimension schließlich wird das zweidimensionale Slab-Gel auf einen speziellen Zylinder gelegt und eine SDS-Gelelektrophorese durchgeführt. Bei diesem Zylinder handelt es sich um einen sieben Zentimeter hohen Gelzylinder aus Acrylamid, mit oberem und unterem Pufferreservoir, alles in 0,1% SDS.
Die dritte Dimension
Was ist mit Konzentration als dritter Dimension gemeint? Oft hat man Zellkulturen, die zunehmenden Konzentrationen einer bestimmten Substanz ausgesetzt werden und man interessiert sich dafür, wie das Proteinexpressionsmuster von der Konzentration abhängt. Ventzki et al. schlagen vor, die Proteine jeder Kultur isoelektrisch zu fokussieren und die Strips dann, schön in Reihe, auf seinen Zylinder zu legen und einer SDS-Gelelektrophorese zu unterwerfen. Das Ergebnis wäre eine 3D-Elektrophorese mit der Substanzkonzentration als dritter Dimension. Entsprechend zeigt die 3D-Elektrophorese mit der Zeit als dritter Dimension, wie das Protein-Expressionsmuster einer Zellkultur von eben dieser abhängt.
Der Ventzki-Zylinder erlaubt auch eine Hochdurchsatz-SDS-Gelelektrophorese: Legen Sie eine Mikrotiterplatte so auf den Gelzylinder, dass die Proteine der Wells auf das Gel geladen werden, können Sie je nach Platte 96, 384 oder gar 1536 Proben gleichzeitig laufen lassen.
Zwei Probleme waren bei der Entwicklung der Methode zu lösen. Zum einen das der ungleichen Hitzeentwicklung im Zylinder während des Laufs, zum anderen das der Probendetektion. Ohne besondere Maßnahmen wird das Innere des Zylinders während der Elektrophorese wärmer werden als das Äußere, weil dort Wärme abgeführt wird. Daher laufen die Proteine im Innern anders als außen. Ventzki et al. lösten das Problem, indem sie den Zylinder isolierten: dadurch erreichen die Ränder die gleiche Temperatur wie das Innere. Abgeführt wird die Wärme durch die auf 10 °C gekühlten Pufferreservoirs oben und unten. Es gibt dann immer noch Temperaturgradienten im Zylinder, sie stehen aber parallel zur Laufrichtung und ein Protein am Rande des Zylinders durchläuft die gleichen Gradienten wie ein Protein in der Mitte.
Wie aber detektiert man die Proben? Ein dreidimensionaler Zylinder lässt sich nicht mit Coomassie oder Silber anfärben, es sei denn, man nimmt sich ein paar Jährchen Zeit dazu. Aber kommen Ihnen nicht schon die Zeiten für das Färben und Entfärben von 2D-Gelen als kleine Ewigkeiten vor? Ventzki et al. markierten die Proteine mit Cy3 und nahmen an einer unteren Ebene des Zylinders die laserinduzierte Fluoreszenz der Proteine mit einer CCD-Kamera auf. Dies solange bis alle Proteine durchgelaufen sind.
Die Methode hat Nachteile: die Apparatur ist kompliziert, die Proben müssen markiert werden und sie gehen beim Lauf verloren, denn sie verschwinden im unteren Pufferreservoir. Man kann die Proteine also hinterher nicht auf dem Massenspektrometer untersuchen.
Dennoch bietet der Ventzki-Zylinder Möglichkeiten: Sie müssen das Gel weder färben noch entfärben und können sich das Ergebnis dennoch gleich nach dem Lauf im Computer anschauen. Und wenn Sie es darauf anlegen, können Sie von 1500 Proben ein SDS-Gel fahren - an einem Tag!
Letzte Änderungen: 09.02.2008