Facelift für das Interface
Flexible HLA-Typisierung
Michael Wittig, Simon Koch, Andre Franke
Wenn Bioinformatiker und Molekularbiologen Softwareprogramme für die Gen-Analyse schreiben, kommt die Nutzerfreundlichkeit meist zu kurz. Warum also nicht Rat beim Industriedesigner holen?
„Gehen ein Molekularbiologe, ein Programmierer und ein Designer zusammen an die Bench..." Dieser Satz klingt fast wie der Anfang eines guten Witzes. Aber selbst wenn er ernst gemeint ist, lässt er nichts Gutes ahnen. Wenn diese unterschiedlichen Disziplinen aufeinander prallen, endet dies meist in einem mehrstündigen und „dreisprachigen“ Meeting. Nichtsdestotrotz fand so ein Treffen tatsächlich statt, und zwar am Institut für Klinische Molekularbiologie (IKMB) der Christian-Albrechts-Universität zu Kiel (CAU). Dort begegneten sich der Programmierer Michael Wittig vom IKMB, der Designer Simon Koch von der Muthesius-Kunsthochschule Kiel und der Molekularbiologe Andre Franke vom IKMB. Ihre Aufgabe: Im Rahmen des EU-Projekts ESGI („European Sequencing and Genotyping Infrastructure“) sollten sie eine hochauflösende Methode zur genetischen Analyse der HLA-Region entwickeln.
Schnell wurde damals klar, dass die existierenden Methoden „von-der-Stange“, die bis dato von Agilent und Raindance vertrieben wurden, gravierende Schwächen im Umgang mit der hochpolymorphen HLA-Region offenbarten. Einige alte HLA-Hasen, darunter auch der Kollaborationspartner aus Oslo (seit mehr als zwei Jahrzehnten im HLA-Geschäft), munkelten schon „...Das schaffen wir doch nie, die Region ist zu komplex...“ Das stachelte den Ehrgeiz der drei Kieler umso stärker an, und so wollten sie jetzt erst recht eine neue NGS-basierte HLA-Analyse-Methode entwickeln.
So wichtig wie Blutgruppen
Aber langsam, was genau ist nochmal die „HLA-Region“? Der humane Leukozytenantigen-Komplex (Human Leukocyte Antigen, HLA) ist die menschliche Version des MHC-Komplexes (Major Histocompatibility Complex) und liegt auf dem kleinen Arm von Chromosom 6. Die vielen Proteine, die durch die Gene in der HLA-Region codiert werden, spielen eine zentrale Rolle in unserem Immunsystem. Kein Wunder also, dass der HLA-Komplex eine „heiße Geschichte“ durchlaufen hat und im Laufe der Evolution durchaus komplex und polymorph geworden ist.
Die HLA-Gruppen sind medizinisch so wichtig wie die Blutgruppen: Viele Schutzimpfungen und Immuntherapien wirken nur bei Personen, die den häufigsten HLA-Gruppen angehören. Das hat einen ganz einfachen wirtschaftlichen Grund: Randgruppen lohnen sich nicht! In Asien haben sich im Laufe der Zeit andere HLA-Gruppen entwickelt als in Europa oder gar im hochdiversen Afrika. Soll auf dem Beipackzettel jeder Impfung angegeben werden, welche HLA-Gruppen ansprechen und welche nicht? Die meisten Laborjournal-Leser sind im Rahmen des Arbeitsschutzes gegen Hepatitis-B geimpft. Es gibt jedoch immer wieder Geimpfte, die seronegativ sind. Experten schätzen, dass etwa 5 % der Geimpften nicht geschützt sind. Bekommen wir daher in Zukunft den personalisierten Impfschutz, passend zu unseren persönlichen HLA-Gruppen?
Die heiße HLA-Geschichte geht aber noch weiter: Fast jede chronisch entzündliche Erkrankung ist mit bestimmten HLA-Allelen und -Loci assoziiert. Bisher konnte man die Rolle von HLA nur bei der Zöliakie aufklären. Klar, nimmt man kein Gluten über die Nahrung auf, so wird man auch die Zöliakie wieder los. Bei sämtlichen anderen Erkrankungen kennt man die genauen Mechanismen jedoch noch nicht, die sich hinter der äußerst starken genetischen HLA-Assoziation verbergen. Morbus Crohn, Colitis Ulcerosa, Psoriasis, Schizophrenie, Typ 1 Diabetes die Liste der HLA-assoziierten Erkrankungen ist äußerst lang. Deswegen sollte in unseren Augen alles daran gesetzt werden, das HLA-Signal für möglichst viele Krankheiten zu „knacken“.
Aber zurück zur „Polymorphie“ der HLA-Gene. Was ist darunter zu verstehen? Das ist sehr einfach: von den HLA-Genen existieren sehr viele verschiedene Varianten (Allele). Als diploider Organismus besitzen wir zwei Allele pro HLA-Gen. Jedes HLA-Protein hat eine Bindungspräferenz für viele verschiedene Peptide, die den Immunzellen im Komplex mit den etwas spezifischeren T-Zellrezeptoren und anderen Faktoren präsentiert werden. Vom HLA-A-Gen sind zum Beispiel mehr als 3.000 Variationen bekannt.
Natürlich hat Homo sapiens diese fein säuberlich in Datenbanken hinterlegt, etwa in der IMGT/HLA-Datenbank des ImMunoGeneTics-Projekts (IMGT; www.ebi.ac.uk/ipd/imgt/hla). Vergleicht man die HLA-Region mit anderen Regionen des menschlichen Genoms, so würde man für Gene dieser Länge nur ein halbes Dutzend verschiedener Allele erwarten. Die hohe genetische Variation der HLA-Gene ist eine von mehreren Grundvorraussetzungen für ein funktionierendes Immunsystem.
Jedes Detail zählt
Man weiß auch, dass heterozygote HLA-Träger ein deutlich geringeres Risiko für viele verschiedene Entzündungserkrankungen haben. Vielleicht, weil hierdurch doppelt so viele verschiedene Peptide gebunden werden können, oder weil homozygote Träger heftiger auf bindende Peptide reagieren. In vielen Fällen des medizinischen Alltags ist es deshalb notwendig, die genauen Allele zu kennen, zum Beispiel als unterstützende Diagnose bei der Zöliakie, beim Morbus Bechterew, und soweiter.
Weitere wichtige Anwendungen der HLA-Typisierung – wir sprechen hier übrigens von einem Multi-Millionen-Geschäft (vielleicht sogar Milliarden) – sind die HLA-Typisierung vor Organtransplantationen (hier muss die Charakterisierung des frisch gestorbenen Organspenders natürlich sehr schnell erfolgen) und für Knochenmarkspenderegister. Gerade letztere typisieren mehrere 1.000 potentielle Spender pro Jahr und dürsten nach schnelleren und billigeren Methoden. Denn nur mit einem großen Schatz an Typisierungsergebnissen kann der perfekte, passende Spender im Notfall aufgespürt werden.
Traditionell werden bei der HLA-Typisierung kleine Stücke des Genoms vervielfältigt und dann mit spezifischen Farbstoff-Sonden überprüft. Aufgrund der Farbstoffreaktion, die ausgelöst wird, wenn eine Sonde ihr Gegenstück bindet, sind Aussagen zu den vorliegenden Allelen möglich. Als Goldstandard gilt jedoch die Sanger-Sequenzierung, die seit mehreren Jahrzehnten etabliert ist. Die Sonden-Methode ist sehr aufwändig, wenn man die unterschiedlichen Allele bis in die DNA-Sequenz typisieren möchte. Für den medizinischen Alltag reicht in der Regel eine niedrige Auflösung der Allel-Sequenz, weil noch zu wenig über die spezifischeren Allele jenseits der Proteinstruktur bekannt ist. Bei Organtransplantationen ist eine höhere Auflösung willkommen, meist beschränkt man sich aber auch hier auf eine niedere Auflösung.
Die Sanger-basierte Typisierung ist zeitaufwändig und in unseren Augen nur unzureichend für den hohen Probendurchsatz von Biobanken oder Blutbanken geeignet. Hinzu kommen Lizenzgebühren für kommerzielle Softwareprodukte. Zudem ist die Amplifikation der DNA, die vor der Sequenzierung nötig ist, fehleranfällig. und die Optimierung der Primer-Sets ist sehr zeitaufwändig. Schneller und günstiger ist die Typisierung mit Next-Generation-Sequenzierern (NGS). Diese Maschinen sind in der Lage, innerhalb weniger Tage ein komplettes Humangenom zu sequenzieren (bei Verbrauchsmittelkosten von derzeit weniger als 1.500 €).
Knackpunkt Genom-Anreicherung
Die HLA-Typisierung mittels NGS verläuft sehr ähnlich wie mit der Sanger-Sequenzierung. Zunächst reichert man den Teil des Genoms, den man ins Auge gefasst hat, mit einer Multiplex-PCR mit mehreren Primern an und sequenziert die Amplikons (Short Amplicon Sequencing). PCRs in polymorphen Abschnitten sind jedoch sehr fehleranfällig und führen immer wieder zu Ausfällen – und daraus resultierend – zur Wiederholung von Experimenten. Interessant ist die Anreicherung mit Hilfe der Long-Range-PCR, aber auch hier sind mehrere parallele PCR-Reaktionen nötig. Die hierzu benötigten Kits kann man kaufen, sie erscheinen uns aber als unangemessen teuer (schließlich sollte die NGS ja günstiger als die Sanger-Sequenzierung sein).
Wir suchten deshalb nach einer Möglichkeit, den kritischen Schritt der PCR-Anreicherung zu umgehen. Dafür bot sich die Targeted Enrichment-Technik an. Bei dieser stellt man RNA-Sonden („Baits”, also „Köder“) her, die den anvisierten DNA-Sequenzen ähneln. Die RNA-Köder versieht man mit Biotin und fischt mit ihnen alle DNA-Segmente aus der Lösung, die der Ziel-Sequenz ähneln. Die Ausfallraten sind hierbei sehr gering, das Verfahren ist aber nicht so spezifisch wie eine PCR.
Im Klartext bedeutet dies, dass man auch sehr viel DNA aus der Lösung fischt, an der man nicht interessiert ist. Das erschwert die Datenanalyse, ist aber ansonsten kein Problem – im Gegenteil. Diese vermeintliche Schwäche der Methode ist gleichzeitig eine Stärke, weil „Ungereimtheiten“ in der Ziel-Sequenz von den RNA-Molekülen toleriert werden. Mit anderen Worten: der richtige DNA-Abschnitt wird angereichert, selbst wenn Insertionen, Deletionen oder SNPs vorliegen (das ist von Vorteil bei Tumorproben!).
Die Technik ist darüber hinaus auch flexibel und skalierbar. Mit zusätzlichen Sonden ist es zum Beispiel möglich, neben der HLA-Typisierung auch andere Regionen des Genoms zu analysieren. In einem aktuellen Experiment typisieren wir auf diese Art nicht nur die HLA-Region, sondern auch gleichzeitig Blutgruppen, Geschlecht, Risikofaktoren für entzündliche Darmerkrankungen und eventuell vorhandene Spuren von Herpes-Viren in den vorliegenden Patientenproben.
Im Detail funktioniert unsere Methode, die wir in diesem Jahr publiziert habe, wie folgt (Wittig et al., Nucleic Acids Research, 1-8). Zuerst extrahiert man die DNA aus einer Blutprobe oder einem anderen Gewebe. Mit Ultraschall oder Enzymen schert man die DNA in kleine Stücke von ca. 200-500 Basenpaaren Länge. Nach der Fragmentierung versieht man die Enden mit kurzen Sequenzen, die die Vervielfältigung der Bruchstücke sowie ihre Sequenzierung auf den NGS-Geräten erlauben.
Kaufen oder selbst synthetisieren
Die so vorbereitete und gegebenenfalls etwas vervielfältigte DNA mischt man im nächsten Schritt unter definierten Bedingungen mit den DNA-bindenden RNA-Sonden. Wir erinnern uns daran, dass diese mit Biotin-Molekülen versehen wurden, die an das Protein Streptavidin binden. Man benötigt also noch kleine magnetische Kügelchen, auf denen das Streptavidin verankert ist. Mit einem Magneten zieht man die gewünschte DNA an den Rand des Gefäßes und pipettiert den Rest aus diesem heraus. Dieses Verfahren ist leicht zu automatisieren und für Pipettierplattformen geeignet.
Die markierten RNA-Köder sind bei verschiedenen Firmen erhältlich oder können selbst synthetisiert werden. Sind die Proben fertig verarbeitet, werden sie mit einem beliebigen NGS-Gerät sequenziert.
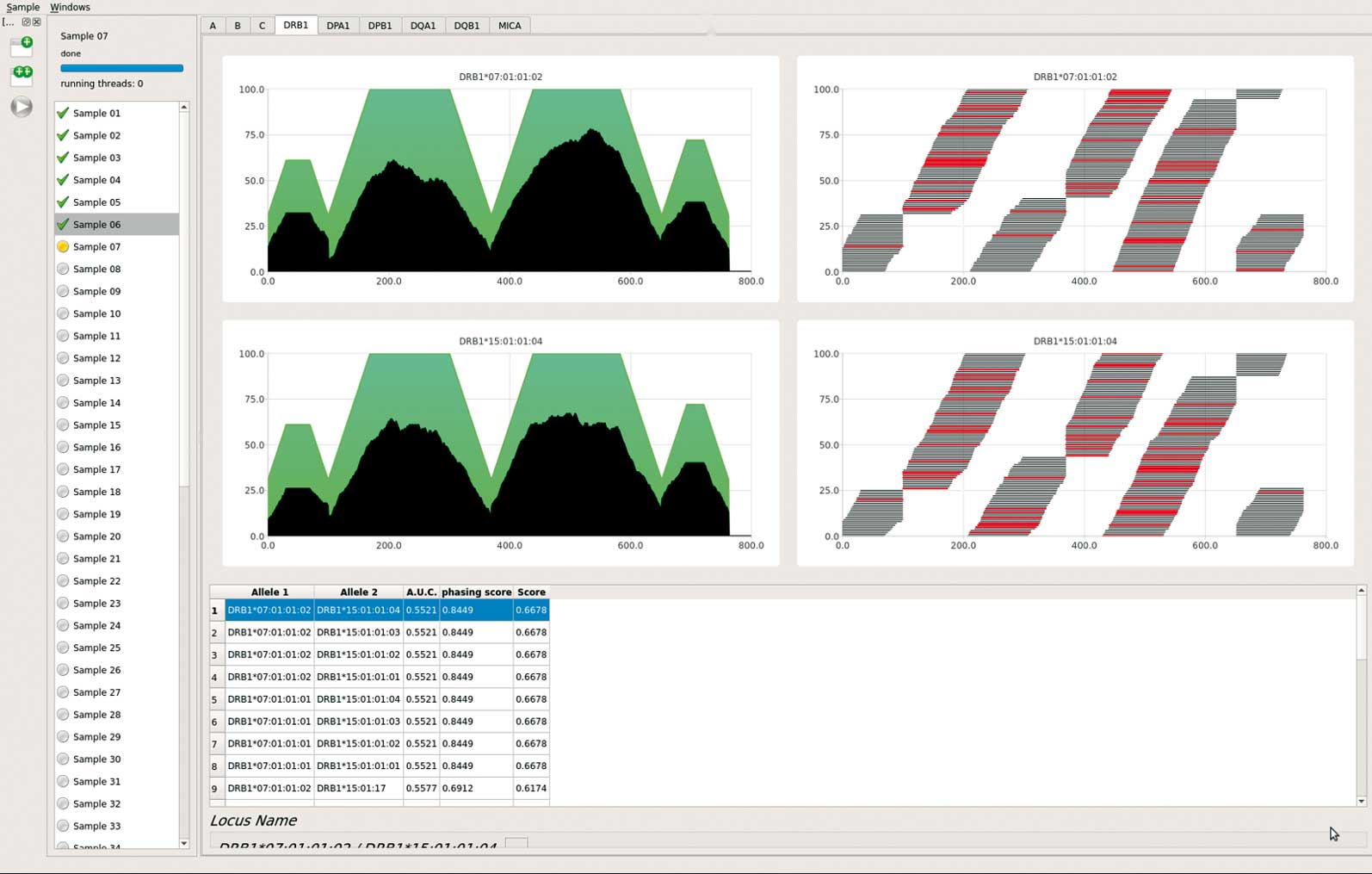
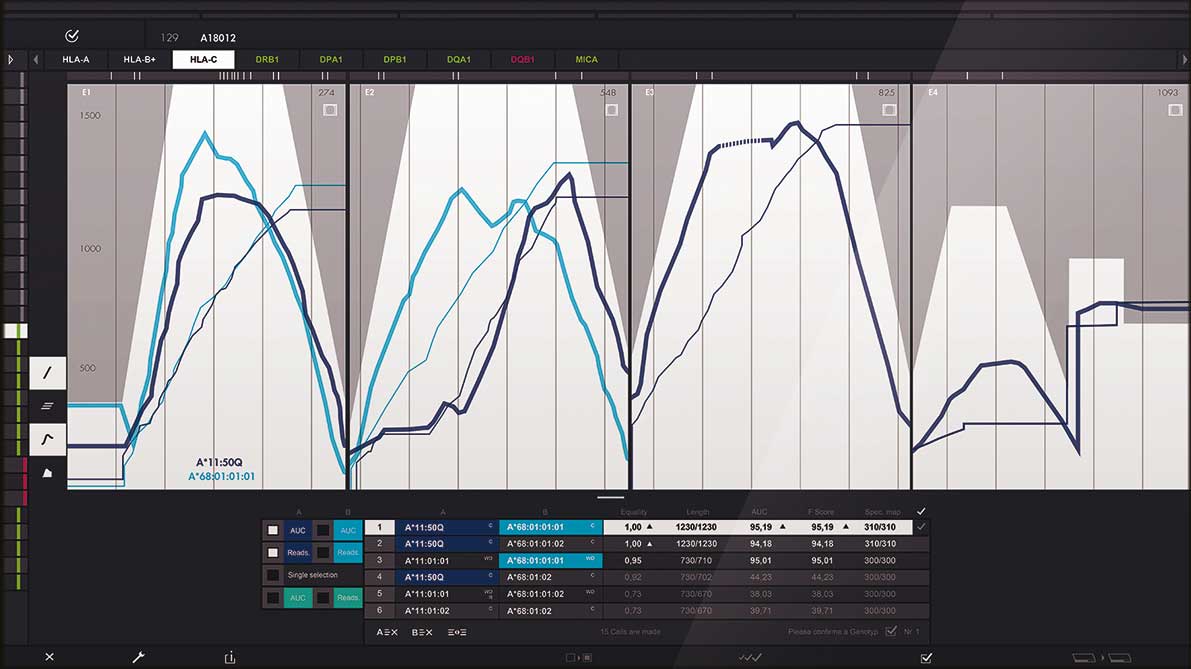
Für die Auswertung der Sequenzdaten haben wir das Programm HLAssign entwickelt, das die Originaldateien des Sequenzierers verarbeitet und fertige Analyseergebnisse liefert. Auf unserer Webseite steht HLAssign zum kostenlosen Download bereit (www.ikmb.uni-kiel.de/resources/download-tools/software/hlassign).
Inzwischen haben wir die Benutzeroberfläche von HLAssign in Zusammenarbeit mit der Muthesius-Kunsthochschule Kiel komplett überarbeitet. Zu Anfang herrschte zugegebenermaßen etwas Skepsis über die Kooperation mit den „Künstlern“. Die drei Mann starke Arbeitsgruppe um Frank Jacob stieg jedoch hochmotiviert in unser Projekt ein und erkannte sofort, dass die erste Oberfläche weder nutzerfreundlich war, noch einem professionellen Design folgte. Es war nun mal ein klassisches Bioinformatik-Programm.
Nur Look and Feel Interface
Nach vielen Entwürfen und Gesprächen mit den Anwendern und dem Programmierer stand nach sechs Monaten das fertige Design. Der Molekularbiologe freute sich, dass er in Kürze ein „durchgestyltes“ und auf den Nutzungskontext hin entworfenes Interface verwenden konnte. Pustekuchen! Mit Schmerzen nahm er zur Kenntnis, dass das Design lediglich ein „Look&Feel“ darstellte und noch lange nicht in der Software implementiert war!
Auch der Informatiker kam ins Straucheln: „Software-Oberflächen in diesem Detail umsetzen? So etwas habe ich noch nicht professionell gemacht, das ist eine eigene Sprache.“ Recht hatte er. Um dem Informatiker beizuspringen: Bioinformatische Anwendungen glänzen meist nicht durch nutzerfreundlich gestaltete Oberflächen. Der Fokus liegt hier meist auf den technischen und methodischen Komponenten. Anwenderfreundlichkeit spielt häufig eine Nebenrolle, wichtiger ist die Erstpublikation.
Die unerwartet hohen Ansprüche des „Künstlers“ an die Gestaltung des Interface, das hierdurch aber erst benutzbar wurde, erhöhten den Aufwand enorm. Aus GUI (Graphical User Interface, die graphische Benutzeroberfläche) wurde ein lautes „Ui“. Die viele Arbeit wurde jedoch belohnt: Simon Koch erhielt für sein Interface Design den iF Student Design Award des iF Industrie Forum Design e.V.
Die Datenanalyse mit der Software funktioniert vereinfacht folgendermaßen. Die oben beschriebene Fragmentierung der DNA verläuft nach einem zufälligen Muster. Dies ist eine wichtige Grundvoraussetzung für den Analysealgorithmus. Die Sequenzen, die das NGS-Gerät liefert (Reads), werden mit einer Kollektion sämtlicher bekannter HLA-Allele (n>10.000) verglichen. Das Programm prüft, auf welche Referenzen die kurzen Reads perfekt passen. Die Reads sind je nach NGS-Lauf meist 100-150 Basenpaare (bp) lang, die Allele bis zu tausend. Im nächsten Schritt ermittelt der Algorithmus die Verteilung der Reads auf die verschiedenen möglichen Genotypen.
Automatische HLA-Typisierung
Somit ist es möglich, mit sehr hoher Wahrscheinlichkeit und voll automatisiert die richtigen Genotypen für die unterschiedlichen HLA-Gene zu bestimmen. Um wirklich auf Nummer sicher zu gehen – und so ist es auch Goldstandard bei der Sanger-Sequenzierung von HLA-Genen – ist es aber weiterhin wichtig, die Daten visuell zu überprüfen. Das Softwaredesign von HLAssign 2.0 unterstützt den Nutzer bei der Entscheidungsfindung.
Mittlerweile wurden am IKMB über 2.000 DNA-Proben mit der beschriebenen Methode typisiert. Natürlich wurde die Technik zuvor an mehreren hundert hochdiversen DNA-Proben validiert. Unter den mehr als 2.000 Proben waren auch 400 DNAs aus der klinischen Versorgung, die in ein Knochenmarkspenderegister geladen und ebenfalls mit herkömmlichen Mitteln validiert wurden. Damit erreichten wir eine erste Translation in die Klinik; weitere Schritte in diese Richtung, zum Beispiel die Akkreditierung der Methode, sind in Vorbereitung.
Am Ende bleibt eine sehr anwendungsnahe Methode und Software, die mit großer Sicherheit weiterentwickelt wird. Außerdem bleibt die Erkenntnis, dass man interdisziplinär häufig mehr erreichen kann als im eigenen Aquarium. Also, liebe Forscherkollegen, verlasst Eure Komfortzonen – manchmal staunt man über die Größe und Vielfalt des Ozeans.
Letzte Änderungen: 30.09.2015