Kampf dem Fehlerteufel
NGS-Datenanalyse mit pibase
Michael Forster & Andre Franke
Next Generation Sequenziergeräte sind schlampige Genies. Und auch auf viele Sequenzanalyse-Programme ist noch zu wenig Verlass.
Auf Fortbildungen und Konferenzen hört man zunehmend vom bevorstehenden Einzug der Next-Generation-Sequenzierung (NGS) in den klinischen Alltag. Viele Klinikleiter, Humangenetiker oder Laborchemiker setzen deshalb schon jetzt ihre Fußsoldaten auf die rechtzeitige Etablierung der NGS-Diagnostik an. Hierbei ist jedoch die Gefahr groß, sich auf einen monatelangen Irrweg zu begeben. Als Selbsterfahrung kann dieser durchaus lehrreich und amüsant sein, aber leider ist er auch äußerst frustrierend.
Für weniger masochistisch veranlagte Leser haben wir die teils schmerzhaften Erfahrungen unserer Gruppe am Institut für Klinische Molekularbiologie der Christian-Albrechts-Universität zu Kiel auf dem steinigen Weg zur Analyse von NGS-Daten zusammengefasst. Zudem erklären wir, wie man mit dem von uns entwickelten Softwarepaket pibase fehlerhafte Punktmutationslisten aus verschiedenen NGS-Programmen vereinigen, transparent gestalten und (teilweise) korrigieren kann (www.ikmb.uni-kiel.de/pibase).
Viele Fallstricke
Um gleich vorweg die grundsätzliche Frage zu beantworten, ob die NGS überhaupt zu etwas taugt: Ja, das tut sie, aber es ist Vorsicht geboten! In unseren eigenen Projekten, sowie anhand von Daten und etablierten Softwareprogrammen aus dem 1000-Genomes-Project, stellten wir auf den ersten Blick Erschreckendes fest. Je nach Datensatz bestimmten die NGS-Analyseprogramme SAMtools und GATK mit den vorgeschlagenen Einstellungen 0,5% bis 60% der relevanten Punktmutationen falsch oder erkannten sie infolge von Sequenzierlücken gar nicht (Li et al., Bioinformatics 25, 2078-9; McKenna et al., Genome Research 20, 1297-03; DePristo et al., Nature Genetics 43, 491-8). Zu ähnlichen Schlüssen kamen auch andere Gruppen (Vogelstein et al., Science 339, 1546-58; O‘Rawe et al., Genome Medicine 5, 28; Lam et al., Nature Biotechnology 1-6). Die Sensitivität und Spezifität der Detektion größerer und komplexerer genomischer Varianten (Insertionen, Deletionen, Kopienzahlvariationen „CNVs“ etc.) ist sogar noch deutlich niedriger als für Punktmutationen, so dass wir diese hier ausklammern.
Als wir die Mutationslisten von GATK, SAMtools oder anderer Programme (alte Life Technology SOLiD Bioscope Versionen, die aktuellste Illumina Casava Version) für Tumor-Normal-Vergleiche von Krebspatienten heranzogen, stellten wir fest, dass eine große Anzahl somatischer Punktmutationen (Unterschiede zwischen Tumor- und Normalprobe) reine Software-Artefakte waren und die Programme viele echte somatische Mutationen nicht anzeigten.
Das EU-ESGI-Konsortium (www.esgi-infrastructure.eu) hat jüngst identische NGS-Rohdaten durch 12 erfahrene NGS-Arbeitsgruppen analysieren lassen. Die Zahl der als „sicher“ detektierten somatischen Punktmutationen lag je nach Arbeitsgruppe zwischen rund 1.000 und 20.000 Mutationen. Der Konsensus zwischen allen Gruppen betrug weniger als 100 Mutationen!
Auswirkung auf Therapie
In einem aktuellen, realen Fall eines Krebspatienten am Universitätsklinikum Schleswig-Holstein, der an dem sogenannten Li-Fraumeni-Syndrom leidet, übersah das Analyseprogramm VARSCAN eine TP53-Mutation mit klinischer Bedeutung für die Therapie, die wir glücklicherweise mit pibase entdeckten. Peter Lichter vom DKFZ in Heidelberg rät in solchen Li Fraumeni-Fällen von einer Bestrahlungstherapie ab, weil das Risiko besteht, dass die Krebszellen nicht zur Apoptose angeregt und gesunde Zellen übermäßig geschädigt werden.
Nun ist die Analyse eines Krebsgenoms – das ja eher eine komplexe Mischung aus vielen abnormalen Einzelgenomen ist – um einiges schwieriger als die Mutationsdetektion in „Normalgenomen“. Aber nicht nur wir beobachteten auch in diesen eine große Zahl falschpositiver (keine echten Mutationen) und falschnegativer (echte Mutationen wurden übersehen) Ergebnisse.
2011 verglich Michael Snyders Gruppe in Stanford die von den Firmen Illumina und Complete Genomics verwendeten Sequenziertechnologien und analysierte die identifizierten Varianten (Lam et al., Nature Biotechnology 1-6 ). Die Schnittmenge der beiden unabhängigen Experimente betrug bei den Einzelpunktvarianten 88,1%. Die Falschpositiv- und Falschnegativraten lagen für Illumina bei 5% beziehungsweise 2%, und für Complete Genomics bei 1% beziehungsweise 5% und betrafen auch klinisch wichtige Punktmutationen. In der Diagnostik strebt man eine Rate von weniger als einem Fehler je 1000 Basenpaare an.
Wie sind diese hohen Fehlerraten zu erklären? NGS-Mutationslisten entstehen in drei Schritten. Im ersten verarbeitet ein Sequenziergerät Millionen oder Milliarden kurzer DNA-Fragmente. Dabei wird typischerweise eine kurze Buchstabensequenz (Basensequenz beziehungsweise „Read“) von jedem Ende des DNA-Fragments abgelesen.
Im zweiten NGS-Schritt berechnet der Computer (in der Regel ein Linux-Rechen-Cluster) den wahrscheinlichsten genomischen Ursprung jedes DNA-Fragments. Dazu gleicht (aligniert) das eingesetzte Analyseprogramm die Sequenzen mit einer Humangenomreferenz ab. Viele DNA-Fragmente lassen sich jedoch nicht nur einer, sondern mehreren Regionen gleichermaßen gut zuordnen. In Zukunft sollen sogenannte exakte Alignments die Trefferquoten hierbei erhöhen. Derzeit sind diese aber noch zu rechenintensiv und müssen für die Analyse kompletter Genomdaten weiter optimiert werden.
Im dritten NGS-Schritt ermittelt man die Mutationen entweder als Varianten zum Referenzgenom oder als somatische Unterschiede zur Keimbahn. Dies ist keineswegs trivial. Auf technologischer Ebene beeinflussen Gerätetyp und Experiment (zum Beispiel Genom-, Exom-, RNA-, oder Targetsequenzierung) die Daten. Auf biologischer Ebene erschweren homologe Regionen im Genom, sowie individuelle Ausprägungen der DNA die Analyse. Hierzu zählen Verluste oder Zugewinne von ganzen Chromosomen oder Segmenten (Ploidie-Änderungen), sowie die Fusion von Segmenten zu Pseudochromosomen (Barker, Cancer genetics and cytogenetics 5, 81-94; Rausch et al., Cell 148, 59-71).
Falsche Vorraussetzungen
Die meisten Programme zur Mutationsermittlung sind jedoch nicht für die Patientendiagnostik entwickelt worden, sondern für Populationsstudien. Bei diesen geht man von einer diploiden und gesunden Bevölkerungsgruppe aus. Je nach Programm und Programmeinstellungen filtert dieses auch echte Alignment- und Sequenzierfehler sowie biologische Mutationen heraus.
Viele NGS-Bioinformatiker versuchen, die Fehlerraten zu verringern, indem sie zwei oder mehr Programme mit Standardeinstellungen verwenden. Die Schnittmenge hieraus soll dann die „sicheren“ Mutationen liefern. Einige Forscher analysieren auch die Vereinigungsmenge aller Mutationslisten, um die Sensitivität zu erhöhen.
Die Genotypisierung mit pibase funktioniert ähnlich. Das Programm verwendet zehn getrennte, interne Genotypisierungsmethoden, die sich durch zunehmend stringentere Qualitätsfilter unterscheiden. Dadurch liefert es nicht nur einen „Konsensusgenotypen“, sondern auch eine Aussage über die Variabilität oder Stabilität dieses Genotypen (variable Genotypen erhalten ein Fragezeichen). pibase zeigt dem Anwender, ob es den Genotyp auf dem Vorwärts- und Rückwärtsstrang erkennt (unabhängige, unterstützende Beobachtungen) oder nur auf einem Strang. Zudem unterscheidet es zwischen Genotypen, die in einer hypervariablen oder homologen Region des Referenzgenoms liegen. pibase eignet sich deshalb zur detaillierten Überprüfung von vorgegebenen Positionen in einem Genom und zur Entscheidung, ob eine vermutete Mutation echt oder falsch ist. Zur Detektion von Mutationen ist es nicht vorgesehen.
Wir haben leidvoll erfahren müssen, dass der herkömmliche Vergleich zweier Proben anhand von Genotypen viele vermeintliche Unterschiede zutage fördert, die sich bei der Validierung als falsch herausstellen. Gleichzeitig übersehen die Programme viele echte Unterschiede. Vor allem unsichere oder vorsorglich „weggefilterte“ Genotypen führen häufig zu Fehlern. Weiterhin bleiben auf dem üblichen Analyseweg grundsätzlich immer Genotypenänderungen auf der Strecke, die mit dem Zugewinn eines Chromosomensegments einhergehen, beispielsweise A/T zu A/T/T.
Griff in die Trickkiste
Um diese Fehler zu vermeiden, greift pibase in die statistische Trickkiste und vergleicht nicht die Genotypen, sondern die zugrunde liegenden Original-Alignmentdateien mit dem Fisher’s Exact Test. Keine zwei Monate nach der Begutachtung unseres pibase-Manuskripts im Jahr 2011 verkündete die VarScan-Homepage, dass man nicht mehr ein heuristisches Verfahren sondern ebenfalls den Fisher’s Exact Test verwende. Ein Zufall wie aus dem wahren Leben. VarScan vergleicht aber lediglich die eindimensionalen Mutationslisten des SAMtools-Programms miteinander. Deshalb übersah es auch die weiter oben erwähnte TP53-Mutation.
Nun fragt man sich als mittelständisches, universitäres Sequenzierzentrum, ob „professionellere“, kommerzielle NGS-Diagnostik-Serviceprovider das Problem der NGS-Datenunschärfe gelöst haben. Zu diesem Zweck schickten wir jüngst zwei Proben (Normal- und zugehöriges Tumorgenom) über den großen Teich zu einer NGS-Firma, die sich den Service auch etwas kosten ließ (7.300 Euro für das Normal- und die doppelte Summe für das Tumorgenom).
Machen es die Profis besser?
Zwar hielt man sich fast an die versprochene – und zugegebenermaßen für uns immer noch beeindruckende – Turn-Around-Time von zwei Wochen für die beiden Genome. Dennoch war die Enttäuschung groß, als die Festplatte mit den Daten in unserem Labor eintrudelte. Zuerst ließ sie sich wegen eines NTFS-Formatfehlers nicht in das Linux-System einbinden (mounten). Nachdem wir dieses Problem, so wie ein weiteres mit einer korrupten MD5-Prüfsumme (mit der man prüft, ob große Dateien Fehler enthalten) gelöst hatten, schauten wir uns den Analysereport und die verwendeten Methoden an.
Auch hier wurden wir enttäuscht, denn der Sequenzierdienstleister hatte weder Kombinationen aus etablierten Tools, noch ausgefeilte, selbst entwickelte Algorithmen verwendet (die oben erwähnte Sequenzier-Firma Complete Genomics setzt zum Beispiel auf eigene, intelligente Algorithmen). Das heißt im Klartext: die von uns beauftragte Firma fütterte ihre Null-Acht-Fünfzehn-Analysesuite, die sie kostenlos mit Ihren Geräten ausliefert, mit Null-Acht-Fünfzehn-Parametern und schickte uns die Daten ungefiltert per Festplatte. Letztlich entpuppte sich das Analyseprogramm als fehlerhaft.
Ihre eigenen Erfahrungen mit der NGS-Sequenzierung und der sich anschließenden Analyse von NGS-Daten können wir Ihnen nicht abnehmen. Mit den nachfolgenden Tipps können Sie jedoch die gröbsten Fehler bei der NGS-Genotypisierung vermeiden:
- Untersuchen Sie alle dbSNP-Positionen in der Original-Alignmentdatei mit pibase.
- Verwenden Sie mehrere Programme, um neue Mutationen aufzuspüren, und überprüfen Sie die Original-Alignmentdatei an diesen Positionen mit pibase.
- Schalten Sie sämtliche Filteroptionen in SAMtools und GATK aus.
- Streben Sie eine Mindestabdeckung (Coverage) von 40fach bei Genomsequenzierungen und mehr für Krebsgenome und Exomsequenzierungen an.
- Sequenzieren Sie die gleiche Probe mit unterschiedlichen Technologien (zum Beispiel Illumina HiSeq vs. Life Technologies SOLiD)
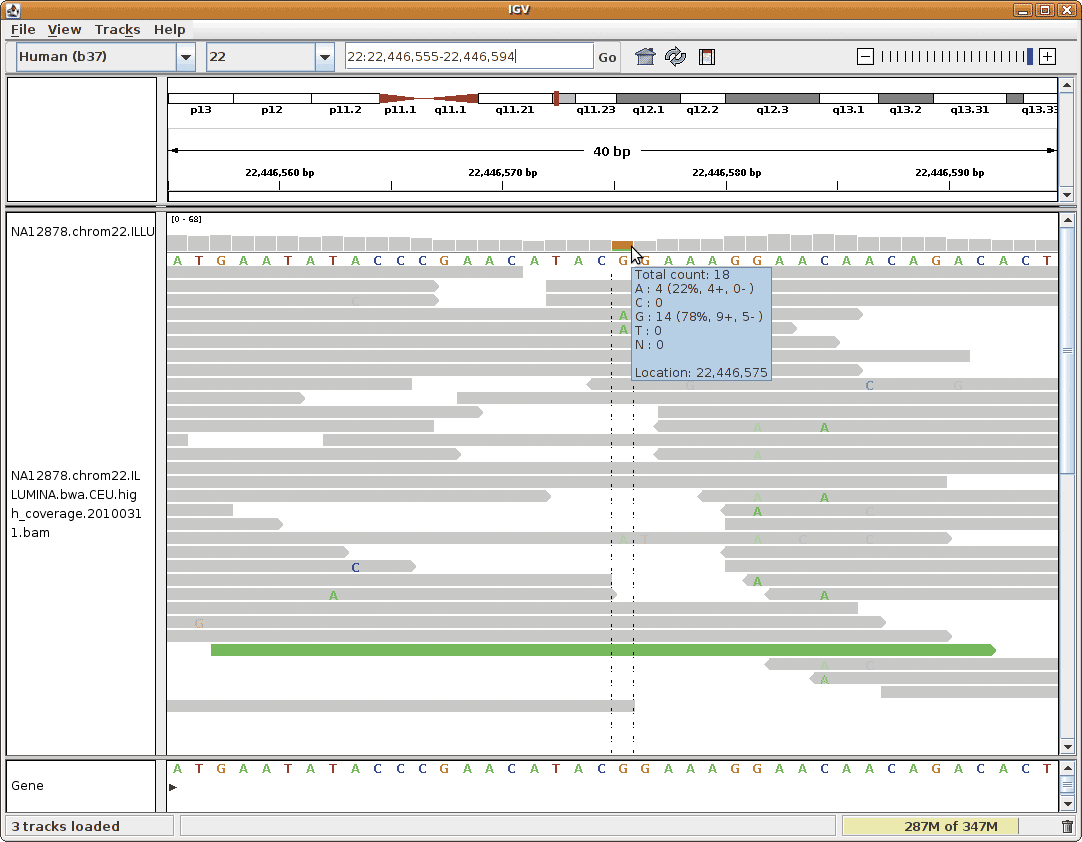
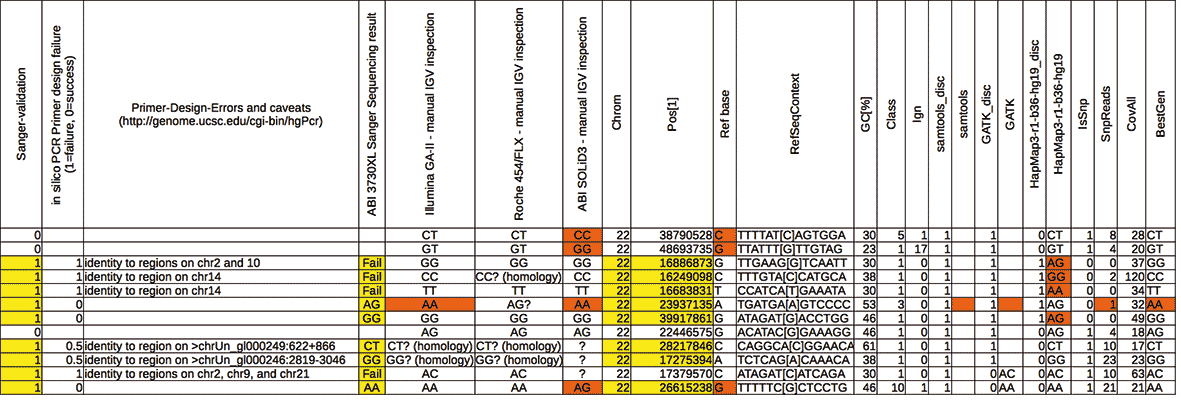
- Verwenden Sie den IGV Viewer zur visuellen Durchsicht der Kandidatenmutationen und validieren Sie kritische Positionen mit der Sanger-Sequenzierung (Robinson et al., Nature Biotechnology 29, 24-6).
- Bedenken Sie, dass auch die Sanger-Sequenzierung für menschliche Fehler anfällig ist (Primerdesign, Interpretation der Elektropherogramme, vertauschte Proben, etc).
- Testen Sie NGS-Programme in einer kontextspezifischen Pilotphase, da diese immer für Überraschungen gut sind.
Letzte Änderungen: 12.06.2013