Live Cell Imaging – Dem Leben zuschauen
Lebende Organismen unter dem Mikroskop zu beobachten hat auch nach über hundert Jahren nichts von seiner Faszination verloren – und immer noch stellt es eine Herausforderung dar.
(15. November 2013) Wer irgendwann mal im Studentenpraktikum vor dem Lichtmikroskop gesessen hat, erinnert sich vielleicht an Pantoffeltierchen, die dauernd aus dem Sichtfeld schwammen. Oder an unscharfe Zellwände durchgeschnittener Tannennadeln. Oder auch an Blutausstriche, in denen es leere Kreise zu bewundern gab, bei denen es sich vermutlich um Erythrozyten handelte – oder um ihre Ghosts.
Am besten erkennen ließen sich Details immer dann, wenn man das Material zuvor fixiert und gefärbt hatte. Präparationstechniken haben in der Mikroskopie eine lange Tradition und ermöglichten überhaupt erst die detailreiche Charakterisierung vieler Strukturen. Man denke etwa an die Chromosomen, deren Name daher rührt, dass sie sich gut anfärben und somit leicht im Lichtmikroskop sichtbar machen lassen. Doch haben eben jene Präparationsmethoden einen entscheidenden Nachteil: Die Zellen werden abgetötet und können schon allein dadurch ihr Aussehen verändern. Hinzu kommen Chemikalien, die die Probe dehydrieren und Proteine denaturieren. Will man dann auch noch Farbstoffe in die Zellen einbringen, müssen diese irgendwie durch die Membran, was in der Regel nicht ohne chemische Gewaltanwendung gelingt. Was man hinterher vor sich hat, mag schön aussehen, unterscheidet sich aber häufig grundlegend von der Situation im lebenden Organismus.
Andererseits: wer ein klassisches Durchlichtmikroskop aus alten Forschertagen im Labor stehen hat, wird in völlig unbehandelten Proben nicht allzu viel erkennen. Glücklicherweise ist die Zeit in der Mikroskopie nicht stehengeblieben, so dass es heute eine Reihe weiterer Möglichkeiten gibt, lebende Zellen mikroskopisch zu beobachten und daraus neue Erkenntnisse zu gewinnen. Eines gibt es aber bislang noch nicht: ein prägnantes deutsches Wort für diese Disziplin namens Live Cell Imaging.
An der Universität Freiburg ist das Know-how rund um die Mikroskopie in einer zentralen Einrichtung gebündelt, die einen besonderen Schwerpunkt auf die Darstellung lebender Zellen legt: Das Life Imaging Center (LIC). 2000 mit einem Sonderforschungsbereich ins Leben gerufen, existiert das Zentrum seit 2008 in seiner heutigen Form und steht Wissenschaftlern aller Fakultäten offen.
Geleitet wird das LIC vom Biologen Roland Nitschke. Ein und dasselbe Objekt über einen längeren Zeitraum hinweg beobachten zu können – darin sieht Nitschke einen bedeutsamen Vorteil beim Mikroskopieren lebender Organismen. Zwar lassen sich Veränderungen auch über die Untersuchung verschiedener Proben zu unterschiedlichen Zeitpunkten dokumentieren, doch manche Fragen kann man auf diesem Wege nicht befriedigend beantworten. Denn fixiert man heute die erste und morgen die zweite Probe, so hat man es ja mit zwei unterschiedlichen Exemplaren zu tun. „Die Wanderung oder Entwicklung einzelner Zellen in einem Embryo beispielsweise können sie damit nicht verfolgen“, so Nitschke. Anders beim Live Cell Imaging: „Der besondere Charme ist doch, dass ich hier alle Stadien an ein und demselben Objekt nacheinander oder sogar an vielen Punkten in einem komplexen Objekt gleichzeitig über die Zeit anschaue.“
Am Anfang war das Daumenkino
Schon 1909 berichtete die New York Times über den Franzosen Jean Comandon, der lebende Trypanosomen, die Erreger der Schlafkrankheit, im Mäuseblut gefilmt hatte. „Damals war das natürlich mehr eine Art Daumenkino“, erläutert Nitschke das Grundprinzip dieser Time-Lapse-Mikroskopie, die später durch die Videotechnik abgelöst wurde. Der VHS-Standard der 1980er Jahre hatte aber eine niedrige Bildqualität. Zudem war man auf 25 oder 30 Bilder pro Sekunde beschränkt und konnte somit auch die Belichtung nicht optimal kontrollieren. „Schwache Lichtsignale wurden über zwei Sekunden summiert, wobei entsprechend aber auch das Hintergrundrauschen anstieg“, erklärt Nitschke. In lebenden Objekten passiert in dieser Zeitspanne so einiges, einzelne Zeitpunkte ließen sich daher nicht scharf abbilden.
Erst mit Beginn des digitalen Zeitalters in den 1990er Jahren revolutionierten CCD-Chips die Foto- und Videotechnik – mittlerweile aus den Life Sciences nicht mehr wegzudenken. So müssen Bilddaten in der modernen Lichtmikroskopie nicht mehr aufwändig auf Zwischenmedien wie Kassetten oder Bildplatten gespeichert werden, sondern landen auf Wunsch direkt im Computer. „Das ermöglicht Aufnahmezeiten im Millisekundenbereich und Bildraten von mehr als 100 pro Sekunde“, schwärmt Nitschke.
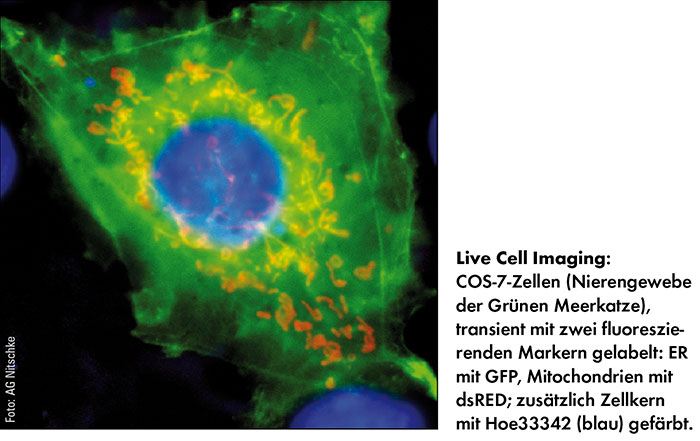
Ebenfalls in den 90er Jahren erlebte die Zellbiologie einen weiteren Quantensprung. Nachdem Douglas Prasher das Green Fluorescence Protein-Gen (GFP) sequenziert hatte, gelang ihm 1994 die Expression dieses Quallen-Proteins in E. coli (Science 1994, 263(5148):802-5). Seither gehört es zum täglich Brot der Genetiker, GFP als Fluoreszenzmarker in Zellen einzubringen. Man setzt die zugehörige DNA-Sequenz einfach hinter den proteinkodierenden Abschnitt des Gens, dessen Produkt man untersuchen möchte. Exprimiert wird dann ein fluoreszierendes Fusionsprotein. Mittlerweile gibt es für den Laboreinsatz eine Flut unterschiedlicher Farbvarianten, die sich aus Fluoreszenzproteinen wie dem GFP ableiten. So lassen sich auch mehrere Proteine gleichzeitig in derselben Zelle mit jeweils einer anderen Farbe markieren und mittels Fluoreszenzmikroskop lokalisieren.
Schädliches Licht
So einfach, wie es sich anhört, ist die Arbeit mit lebenden Zellen dann aber doch nicht. Ein Problem ist das Licht. „In der Intensität, wie man es in der Mikroskopie verwendet, ist es häufig sehr schädlich für einen lebenden Organismus“, erklärt Nitschke. Bedient man sich der Weitfeldmikroskopie, wird die gesamte Probe angestrahlt. Die Arbeit mit Fluoreszenzfarbstoffen hat den Vorteil, dass man sich für die Beleuchtung auf einen schmalen Wellenlängenbereich beschränken kann und so die Zellen schont – sollte man meinen. „Jedoch sind die zu detektierenden Fluoreszenzsignale mindestens 10.000-fach schwächer als die Anregungslichtintensität“, schränkt Nitschke ein.
Bleibt noch der Ausweg, nicht die gesamte Probe zu bestrahlen und beispielsweise ein Konfokalmikroskop zu verwenden. Dabei wird das Objekt gerastert und jeder Bildpunkt nur im Moment der Aufnahme in der Fokusebene belichtet. „Aber die Lichtintensitäten in diesem Punkt sind dann deutlich höher als bei einem konventionellen Lichtmikroskop“, führt Nitschke aus. Denn die Lichtsignale werden innerhalb von Mikrosekunden aufgezeichnet, weshalb die Anregungsintensitäten entsprechend hoch sein müssen, um in diesem winzigen Zeitfenster eine detektierbare Fluoreszenz zu bekommen. „Da kann man schon nach wenigen Bildern Apoptose in Gang setzen“, so Nitschke. Zudem seien auch die Fluoreszenzproteine selbst nicht ohne Nebenwirkungen. „Es entstehen Synthesewege in der Zelle, um das Protein herzustellen, die auf Kosten anderer Prozesse gehen können.“ Dabei, ergänzt er, könnten auch toxische Nebenprodukte entstehen.
Als gewebsschonende Technik gilt die Zwei-Photonen-Mikroskopie. Dabei wird energiearmes langwelliges Licht verwendet, um Fluorophore in der Zelle anzuregen. Der Trick: Es müssen zwei Photonen gleichzeitig auf das Molekül treffen, damit die Anregungsenergie groß genug ist, und dies geschieht fast nur in der Fokusebene. Ähnlich der Konfokalmikroskopie wird dabei die Probe Punkt für Punkt abgescannt, das Gewebe insgesamt jedoch durch das energieärmere Licht geschont. „Die Schäden in der Fokusebene darf man trotzdem nicht vernachlässigen, und die sind bei der Zwei-Photonen-Anregung lokal sogar noch größer als in der konfokalen Fluoreszenzmikroskopie“, bremst Nitschke die Erwartungen.
Selbst für unfixierte Lebendproben gilt also: Bei jedem Blick durch das Mikroskop sieht man zwangsläufig einen Zustand, der sich von den natürlichen Bedingungen unterscheidet. Wie kann man also sicherstellen, dass man einen repräsentativen Eindruck von Organismen bekommt, die man über Stunden oder gar Tage im Mikroskop beobachten möchte? Zunächst einmal sollten die Proben so wenig wie möglich einer Belichtung ausgesetzt werden. Will man etwa dokumentieren, was mit einem bestimmten Protein kurz vor der Zellteilung passiert, wird man viele Aufnahmen in wenigen Minuten anfertigen. Beobachtet man hingegen das Wachstum einer Arabidopsis-Wurzel, sollte man größere Zeitintervalle wählen und die Gesamtdosis des Lichts damit auf mehrere Stunden bis Tage verteilen. Nitschke mahnt, die Beobachtungen stets mit Vorsicht zu interpretieren. Zerstörte Zellen seien dabei das kleinere Übel. „Was stirbt, das kann ich sehen, aber wenn sich ein Organismus einfach nur anders verhält, dann bemerke ich vielleicht gar nicht, dass etwas nicht stimmt.“
Schon mit einfachen Zellkulturen machte Nitschke die Erfahrung, dass sich bereits bei wenigen hundert Bildern die Teilungsrate deutlich verringern kann. In C. elegans kann zu viel Licht im Konfokalmikroskop zu Störungen bei der Entwicklung führen. „Dann entwickeln sich bestimmte Neuronenpopulationen nicht mehr“, warnt Nitschke. Er empfiehlt daher, vor Beginn einer Versuchsreihe zunächst zu testen, ob sich die Ergebnisse in Abhängigkeit von der Lichtexposition unterscheiden. „Verhalten sich meine Zellen ganz anders, wenn ich statt 50 Bildern 1.000 Bilder am Tag mache, sollten die Alarmglocken schrillen!“
Gehirne in 3D
Wer über Stunden oder gar Tage ein und dasselbe Objekt unter dem Mikroskop liegen hat, steht noch vor einer ganz anderen Herausforderung: Unter dem Objektiv müssen Zellkulturbedingungen herrschen. „Entweder rüste ich mein Mikroskop mit einem geeigneten Tisch aus oder ich platziere es im Inkubator“, bringt es Nitschke auf den Punkt. Für beide Varianten ist das Freiburger Life Imaging Center ausgerüstet und bietet Möglichkeiten, wovon die meisten molekularbiologischen Institute nur träumen können. So wundert es nicht, dass Nitschke seit einigen Jahren kaum mehr eigene zellbiologische Projekte verfolgt, sondern mit der methodischen Betreuung der Gastwissenschaftler am LIC ausgelastet ist. So steht denn auch sein Name auf Papern zu den unterschiedlichsten Themen: Mikrotubuli-Dynamik, diverse Signalwege oder auch das Wachstum von Pflanzenwurzeln. Allen Publikationen gemeinsam sind die bunten Bilder, auf denen es irgendwo rot oder grün leuchtet.
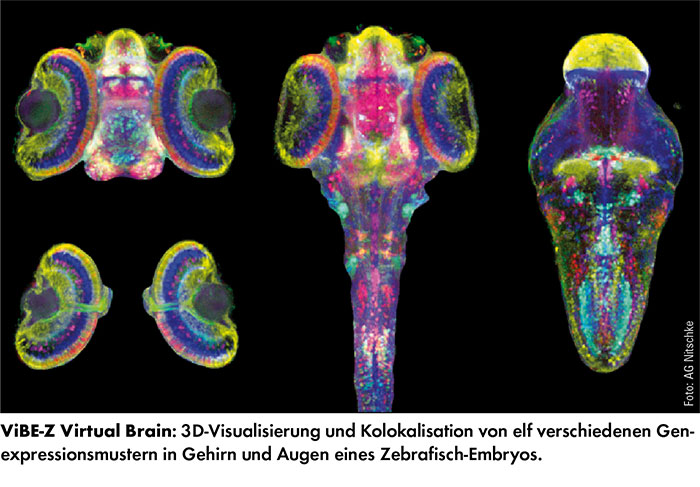
Außerdem arbeitet Nitschke bei der Entwicklung neuer Methoden mit Firmen wie Zeiss oder ibidi zusammen. Dabei geht es nicht nur um Mikroskope, sondern auch um die Verarbeitung der Bilddaten am Rechner. Eine Herausforderung ist das Mikroskopieren dreidimensionaler Objekte. Denn je tiefer man beispielsweise in einen Fischembryo hineinfokussiert, desto schlechter wird die Bildqualität. Diesen Qualitätsverlust vermindert Nitschke zusammen mit Entwicklungsbiologen und Informatikern in einem Projekt namens ViBE-Z (Virtual Brain Explorer in Zebrafisch) mit einem Trick. Bei ViBE-Z geht es um die Gehirnentwicklung des Zebrabärblings und die vergleichende Auswertung der Expression diverser Gene (Nat Methods 2012, 9(7):735-42).
„ViBE-Z kann man mit einem konfokalen Mikroskop von der Stange machen. Wir haben dafür eine spezielle Halterung entwickelt, mit der man das Objekt präzise um 180 ° drehen kann“, erläutert Nitschke. So entstehen von tieferen Schichten doppelte Bilder, eins von oben und eins von unten aufgenommen. Jedes für sich genommen ist suboptimal, beide zusammen aber enthalten ausreichend Information, um daraus ein gutes Bild berechnen zu können. „Herausgekommen sind 3D-Atlanten von Zebrafisch-Gehirnmodellen mit 17 verschiedenen Expressionsmustern. Dieses methodische Grundprinzip lässt sich auch auf andere Organismen anwenden. Zwar wurden die Embryonen für diese Studie abgetötet, doch Nitschke möchte nun ein ähnliches Verfahren auch für die Lichtscheibenfluoreszenzmikroskopie (Lightsheet Fluorescence Microscopy, LSFM) etablieren – und so auch lebenden Organismen bei der Entwicklung zuschauen.
Generative Modelle
Ebenfalls an der Freiburger Uni arbeitet die Medizinerin und Zellbiologin Melanie Börries zusammen mit dem auf Bioinformatik spezialisierten Physiker Hauke Busch. Sie kooperieren seit mehreren Jahren eng mit dem Computerbiologen Robert Murphy, der für seine Projekte zwischen Pittsburgh und Freiburg pendelt. Die drei Forscher wollen wissen, wie Differenzierungsprozesse von Nervenzellen ablaufen und schauen sich hierzu lebende Zellkulturen an. Zum Mikroskopieren gehen sie ins LIC, die weitere Auswertung und Bildanalyse findet dann am Computer statt. Ziel ist es, generative Modelle für Entwicklungsprozesse zu erstellen. Was es damit auf sich hat, erläutert Murphy anhand eines Beispiels: „Als Kind wurden uns Zahlen und Buchstaben gezeigt, beispielsweise viele verschiedene Exemplare einer Fünf. Doch wir haben diese nicht einfach auswendig gelernt, sondern ein generatives Modell verinnerlicht, dass es uns erlaubt, neue Fünfen zu erschaffen.“
Analog möchte Murphy nicht einfach nur wissen, wie sich eine einzelne Zelle differenziert, sondern ein allgemeingültiges Modell für diese Prozesse ermitteln. Dieses Ziel hat jeder Entwicklungsbiologe, doch die Methodik ist eine andere. Klassischerweise würde man sich repräsentative Zellen herauspicken und deren Veränderungen über die Zeit dokumentieren. Doch genau darin sieht Börries ein Problem: „Wenn ich mir nur die beste Zelle anschaue und über die Zeit verfolge, sehe ich dann wirklich das, was auch in den anderen Zellen abläuft?“, fragt sie. „So ist es nämlich nicht, tatsächlich sind die Prozesse in jeder Zelle etwas unterschiedlich.“ Ein generatives Modell würde nun beantworten, was in einer Zelle generell passieren muss, damit bestimmte Prozesse anlaufen.
xxxx
Für ihr aktuelles Projekt verwenden die drei Wissenschaftler eine PC12-Zelllinie (abgeleitet aus einem Phäochromozytom des Nebennierenmarks einer Ratte). Durch Zugabe von Nervenwachstumsfaktor (NGF) differenzieren diese zu Nervenzellen. Die Auswahl beim Mikroskopieren erfolgt zufällig, um nicht einfach die augenscheinlich schönsten Zellen herauszupicken. Alle 12 Stunden werden 100 Zellen aufgenommen. Am Computer soll daraus nun eine Art ideale Zelle errechnet werden. Dabei werden einzelne Zellorganellen jeweils als eigenes Modell dargestellt. So gibt es ein Modell für die Mikrotubuli, ein Modell für die Form des Zellkerns oder ein Modell für die Form der Zelle. Diese Einzelmodelle bauen aufeinander auf und werden zu einem Gesamtmodell der Zelle vernetzt. Hinzu kommen Transkriptomdaten, die zu den einzelnen Zeitpunkten für jedes Gen erhoben werden. „Das machen wir mit Microarrays“, so Busch.
Bislang sind die Ergebnisse nicht publiziert, denn die Studie läuft noch. Doch schon jetzt gibt es ein paar Überraschungen. Denn eigentlich ging man davon aus, dass vor allem in den ersten Stunden nach NGF-Zugabe Signalwege in der Zelle anlaufen, die die Differenzierung steuern. „Wir dachten immer, dass nach 12 bis 16 Stunden der Käse gegessen sei“, so Busch, „das Modell zeigt uns aber, dass anscheinend auch zu späteren Zeitpunkten noch Umprogrammierungen stattfinden, sogar nach 36 oder 48 Stunden.“
Doch woher weiß man, dass dieses Modell valide ist und realistische Daten ausspuckt? „Alle Modelle sind erst einmal per se falsch“, scherzt Murphy und betont dabei, dass man es ja prinzipiell mit Vereinfachungen und Idealisierungen zu tun habe. Um einen ersten Eindruck zu bekommen, wie zuverlässig ein Modell ist, gibt es einen einfachen Weg, so Murphy: „Sie haben eine bestimmte Menge an experimentellen Messdaten, wovon Sie nur einen Teil verwenden sollten, um Ihr Modell zu konstruieren. Dann schauen Sie, ob sich das, was Ihr Modell vorhersagt, mit den zurückgehaltenen Daten deckt.“ Später müsse man dann natürlich schauen, ob weitere Experimente die Vorhersagen bestätigen, die sich aus einem Modell ergeben.
Die Zeit anhalten
Zwar geht es beim Live Cell Imaging vor allem um Lichtmikroskopie, doch wäre es nicht faszinierend, einen elektronenmikroskopischen Blick auf Vorgänge in lebenden Zellen zu werfen, etwa einem Membranprotein bei der Arbeit zuzuschauen? Leider gibt es da eine Hürde: Irdisches Leben hat ein Problem mit Vakuum, ohne Luftdruck würde flüssiges Wasser sofort verdampfen. Genau ein solches Vakuum aber herrscht in einem Elektronenmikroskop. Dem sollen nun ultradünne Kammern standhalten, die oben und unten von einer jeweils 50 nm dicken Scheibe aus Siliziumnitrid zusammengehalten werden und ein Volumen eingrenzen, in dem Viren in wässriger Umgebung beobachtet werden können (Lab Chip 2013, 13(2):216-9).
Henning Stahlberg jedoch zweifelt daran, dass diese Kammern einen echten Nutzen bringen, wenn man lebende Zellen untersuchen möchte. Er leitet eine Arbeitsgruppe am Basler Biozentrum und arbeitet sowohl licht- als auch elektronenmikroskopisch. Abgesehen vom Vakuum sieht er ein weiteres Problem für In vivo-Studien im Elektronenmikroskop. „Die Proben werden wortwörtlich vom Blitz getroffen“. Da eine Zelle diese Prozedur nicht lange überlebt, müsse man das Foto sehr schnell im Kasten haben. „Mit einem Kurzzeitblitz von wenigen Femtosekunden kann man das hinbekommen“, so Stahlberg, „aber außer im Falle von speziellen Fragestellungen sehe ich keinen Grund, diese Proben überhaupt erst in flüssigem Wasser zu haben.“
Stattdessen friert Stahlberg seine Bakterien ein, wenn er die Situation am lebenden Objekt beurteilen möchte. „Wenn ich das innerhalb einer zehntausendstel Sekunde mache, hat das Wasser keine Zeit, Eiskristalle zu bilden“, erklärt er. Diese würden nämlich die Zelle zerstören. Durch das Schockgefrieren hat man aber eine Art Standbild der lebenden Zelle. „Die Zeit bleibt quasi stehen“, so Stahlberg, und für Bakterien funktioniere das sehr gut. Auf diese Weise hat er zusammen mit Fachkollegen die Struktur einer molekularen Injektionsnadel untersucht, die in der Membran von Yersinia enterocolitica sitzt (eLife 2013:e00792). Mit dieser Nadel injizieren die Erreger Moleküle in ihre Wirtszellen, um sie zu ihren Gunsten umzuprogrammieren.
Um Kontrast und Signal-Rausch-Verhältnis zu erhöhen, verwendet Stahlberg keine Schwermetallatome – dies würde eine chemische Fixierung erfordern und den nativen Zustand der Bakterien zerstören –, sondern er errechnet am Computer aus mehreren Aufnahmen des Proteinkomplexes ein einheitliches Bild. Da die Proben gekippt und aus mehreren Winkeln aufgenommen werden, entstehen am Ende dreidimensionale Abbildungen (für Details siehe Artikel „Elektronenmikroskopie – Methode der Wahl“ ab S. 42).
Eukaryotische Zellen würde Stahlberg aber nicht schockgefrieren, denn durch ihr größeres Volumen kühlen sie langsamer ab, so dass sich im Inneren dann doch Eiskristalle bilden können. Tierisches Probenmaterial friert er unter hohem Druck ein, um die Eiskristallbildung zu vermeiden.
Interdisziplinär
Für Stahlberg sind Elektronen- und Lichtmikroskopie keine unvereinbaren Welten. Er kombiniert sie sogar und verrechnet Fluoreszenzbilder mit solchen, die Unterschiede in der Elektronendichte zeigen – „eine Methode, welche als Correlative Light and Electron Microscopy (CLEM) in vielen Laboren weltweit vorangetrieben wird“, ergänzt er. Verschiedene Zeitpunkte in ein und derselben Zelle lassen sich damit aber nicht verfolgen. Denn selbst wenn man lebende Organismen im Elektronenmikroskop hätte – mit der ersten Aufnahme fänden alle zellulären Prozesse ein Ende.
Wollte man ein Schlagwort zum Thema Live Cell Imaging im neuen Jahrtausend finden, so wäre wohl „interdisziplinär“ die treffendste Vokabel. Denn heute geht es dabei um weit mehr als die Arbeit mit dem Mikroskop. Vor allem moderne Computer und der Einsatz intelligenter Steuerungssysteme und Algorithmen eröffnen vollkommen neue Möglichkeiten. Sei es die Kombination verschiedener Bilddaten, die Vereinigung von Licht- und Elektronenmikroskopie oder das Zusammenführen mit Omics-Daten.
Hätte Jean Comandon eine Zeitmaschine, würde er in diesen Tagen sicher mal in Freiburg oder Basel vorbeischauen.
Mario Rembold
(Der Artikel erschien bereits in print im Special "Mikroskopie in der Zellbiologie", Laborjournal 11/2013, S. 47-50; Illustration: Chris Schlag))
Letzte Änderungen: 05.01.2014




